Cardiology
A Chapter in Core Concepts of Pediatrics, 2nd Edition
Ashraf Aly MD, PhD and Soham Dasgupta MBBS
|
The authors want to acknowledged the contribution of Dr. Vidit Bhargava for assistance with image creation. |
|
|
|
||||||||
|
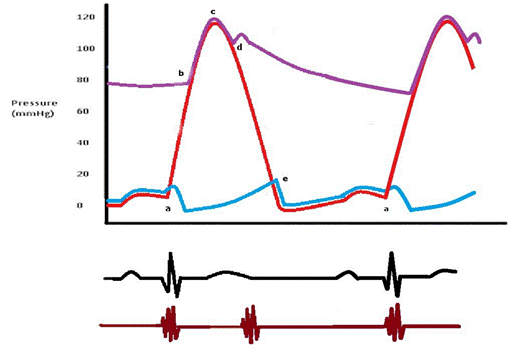
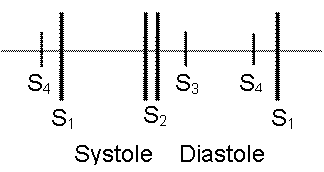
Figure 1. Cardiac cycle of the left side of the heart. The EKG below the diagram shows the corresponding waves with each phase of the cardiac cycle. The bottom line represents the first and second heart sounds. |
|||||||||
The cardiac cycle represents the hemodynamic and electric changes that occur in systole and diastole. It has many phases.
The first heart sound (S1) represents closure of the atrioventricular (mitral and tricuspid) valves as the ventricular pressures exceed atrial pressures at the beginning of systole (point a). S1 is normally a single sound because mitral and tricuspid valve closure occurs almost simultaneously. Clinically S1 corresponds to the pulse.
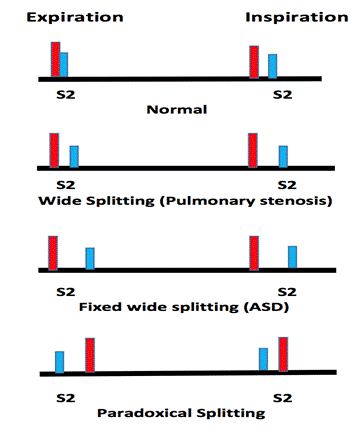
The second heart sound (S2) represents closure of the semilunar (aortic and pulmonary) valves (point d). S2 is normally split because the aortic valve (A2) closes before the pulmonary valve (P2). The closing pressure (the diastolic arterial pressure) on the left is 80 mmHg as compared to only 10 mmHg on the right. This higher closing pressure leads to earlier closure of the aortic valve. In addition, the more muscular and stiff "less compliant" left ventricle (LV) empties earlier than the right ventricle. The venous return to the right ventricle (RV) increases during inspiration due to negative intrathoracic pressure and P2 is even more delayed, so it is normal for the split of the second heart sound to widen during inspiration and to narrow during expiration. Clinically, this is more remarkable with slow heart rates.
Abnormally wide splitting of S2 may occur in :
a) RV volume overload, such as atrial septal defect (ASD). In that case, the split is usually wide and fixed with no change difference between inspiration and expiration due to fixed RV volume (see ASD section).
b) RV outflow obstruction, such as pulmonary stenosis.
c) Delayed RV depolarization such as complete right bundle branch block.
Narrow splitting of S2 occurs in:
a) Pulmonary hypertension as the pulmonary valve closes earlier due to high pulmonary resistance.
b) Mild to moderate aortic stenosis as the A2 is delayed.
Single S2 may occur in:
a) If one of the semilunar valves is missing, as in pulmonary or aortic valve atresia and truncus arteriosus.
b) If both valves close simultaneously as in double outlet single ventricle or in large VSD with equal ventricular pressures
c) In pulmonary hypertension with equal right and left ventricular pressures.
Paradoxical splitting of S2 (P2 is heard before A2) occurs in:
a) Severe aortic stenosis
b) Left bundle branch block
In both conditions, the aortic valve (A2) closes after the pulmonary valve (P2). Since the respiration only affects P2, its effect in paradoxical splitting is the opposite of normal, i.e. inspiration causes narrow splitting while expiration causes wide splitting of S2
The third heart sound (S3) represents a transition from rapid to slow ventricular filling in early diastole. S3 may be heard in normal children.
The fourth heart sound (S4) is an abnormal late diastolic sound caused by forcible atrial contraction in the presence of decreased ventricular compliance.
|
|
|
|
:A2
|
:P2
|
|
A figure showing normal and abnormal splitting of the second heart sound |
|
Murmurs are additional sounds generated by turbulent blood flow in the heart and blood vessels. Murmurs may be systolic, diastolic or continuous.
Systolic murmurs are the most common types of murmurs in children and based on their timing within systole, they are classified into:
a) Systolic ejection murmurs (SEM, crescendo-decrescendo) result from turbulent blood flow due to obstruction (actual or relative) across the semilunar valves, outflow tracts or arteries. The murmur is heard shortly after S1 (pulse). The intensity of the murmur increases as more blood flows across an obstruction and then decreases (crescendo-decrescendo or diamond shaped). Innocent murmurs are the most common cause of SEM (see below). Other causes include stenotic lesions (aortic and pulmonary stenosis, coarctation of the aorta, tetralogy of Fallot) or relative pulmonary stenosis due to increased flow from an ASD
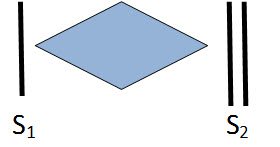
Crescendo decrescendo murmur
b) Holosystolic (regurgitant) murmurs start at the beginning of S1 (pulse) and continue to S2. Examples: ventricular septal defect (VSD), mitral and tricuspid valve regurgitation.
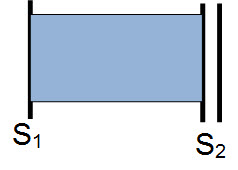
Holosystolic mumur
c) Decrescendo systolic murmur is a subtype of holosystolic murmur that may be heard in patients with small VSDs. In the latter part of systole, the small VSD may close or become so small to not allow discernible flow through and the murmur is no longer audible.
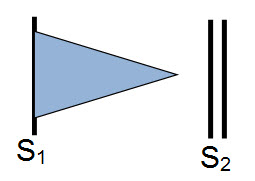
Decrescendo murmur
Diastolic murmurs are usually abnormal, and may be early, mid or late diastolic.
|
More information: Examples of innocent murmu |
|
Continuous murmurs are heard during both systole and diastole. They occur when there is a constant shunt between a high and low pressure blood vessel. Examples: patent ductus arteriosus (PDA) and systemic arterio-venous fistulas. This may also occur in surgically placed shunts such as a BT shunt between the aorta and the pulmonary artery.
Innocent murmurs are common in children and have the following characteristics:
| Table showing the common systolic, diastolic and continuous heart murmurs | |
Systolic |
|
Diastolic |
|
Continuous |
|
| *Obstructive lesions include AS, PS, Coarctation of the aorta, TOF, etc. | |
| Table showing the common heart murmurs audible at different age | |
|
Immediately after birth |
PDA or obstructive lesions* |
|
Shortly after birth (a few hours to few weeks) |
VSD, PDA, PPS (peripheral pulmonary stenosis) |
|
1-4 years |
Innocent murmurs, ASD |
|
Teenage |
Innocent murmur, HOCM or MVP/MR |
| *Obstructive lesions include AS, PS, Coarctation of the aorta, TOF, etc | |
The flow through the systemic and pulmonary circulations is normally balanced and equal in volume (Qp/Qs =1). The two circulations are placed in series with each other. The same volume of blood first makes its way through the systemic circulation, then the pulmonary circulation, then back to the systemic circulation, and so on.
Left to right shunts are characterized by a "back-leak" of blood from the systemic to the pulmonary circulation. This causes the pulmonary flow to be larger than the systemic flow (Qp/Qs >1). As a consequence, the pulmonary circulation carries not only the blood that entered the right atrium and right ventricle through the superior and inferior vena cava, but also the additional blood entering through a VSD, ASD, AVSD or PDA. Blood volume and/or pressure in the pulmonary circulation become abnormally high. If the shunt is significant, there is progressive damage to the pulmonary vasculature and gradual development of irreversible pulmonary hypertension. The pressure in the pulmonary circuit may ultimately exceed the systemic pressure causing reversal of blood flow from the right side of the circulation to the left (Eisenmenger syndrome). This may take as short as 1-2 years in a large VSD, AVSD or PDA or as long as a few decades as in ASD.
Lesions resulting in left to right shunts include:
In small (restrictive) VSD and PDA, the direction and magnitude of the shunt depends on the pressure difference across the shunt. In large VSD and PDA, the direction and magnitude of the shunt depends on the relative resistance in the pulmonary and systemic circuits.
In ASD, the magnitude of the shunt depends largely on relative ventricular compliance (elasticity).
AVSD is a mix between ASD and VSD depending on the predominant lesion.
The defect size is determined by comparing its diameter with the aortic annulus. A small defect is less than 1/3 of the aortic annulus. A moderate sized defect is 1/3 to 2/3 and a large defect is >2/3 the size of the aortic valve annulus
Isolated ventricular septal defects (VSDs) constitute 25-30% of all congenital heart diseases (CHD) in children. VSD may be present in 50% of CHDs such as in tetralogy of Fallot, double outlet right ventricle, truncus arteriosus and others.
Approximately 70% of all VSDs are present in the membranous portion of the inter-ventricular septum, about 20% are in the muscular portion, and the remaining defects are at either the inlet or the outlet portions of the ventricular septum. The inlet VSD is usually a part of atrioventricular septal (AV canal) defect. The outlet (supra-cristal) VSD is more common (about 20%) in the oriental population (or East Asian population).
With a small sized VSD, "restrictive VSD," the direction and magnitude of the shunt depends on the size of the VSD and the pressure gradient between the left and right ventricles. The restrictive nature of the VSD maintains the pressure gradient between the two ventricles.
With a large VSD, the hole is not restrictive and the pressure in both ventricles is almost equal. The direction and magnitude of the shunt depend on the relative difference between the pulmonary and the systemic vascular resistances. In fetal life, the pulmonary resistance is higher than the systemic resistance. As the lungs expand with the first breath, the pulmonary resistance drops significantly and the pulmonary flow increases. The pulmonary resistance continues to decrease until reaching the normal adult ratio of 1:10 by 4-8 weeks (see figure).
Prolonged large left-to-right shunt leads to gradual increase in the pulmonary pressure and pulmonary hypertension eventually develops. As the pressure difference between the systemic and pulmonary systems decreases, the flow across the shunt also decreases. If the pulmonary vascular resistance exceeds the systemic vascular resistance, the direction of the shunt reverses and cyanosis develops (Eisenmenger syndrome). This may develop within two years in otherwise healthy kids and within one year in patients with Down's syndrome.
Children with a small VSD are usually asymptomatic. An incidentally discovered holosystolic or decrescendo heart murmur is the most common presentation. The murmur is commonly discovered at 2-4 weeks of age as the pulmonary vascular resistance drops and the pressure difference between the two ventricles becomes remarkable (Figure). The intensity of the murmur is inversely proportional to the size of the VSD because of the increased turbulence and flow velocity produced by a smaller defect. A thrill may be palpable in some cases. The defect may be small enough to almost close at the end of a systolic decrescendo murmur.
An infant with a large VSD may be asymptomatic in the first few days/weeks of life until the pulmonary vascular resistance drops. As the pulmonary resistance decreases, the left-to-right shunt increases. The right ventricle is thus subjected to high pressure and becomes hypertrophied while the left atrium and left ventricle receive more volume and become dilated. The right atrium is not usually affected. Congestive heart failure (CHF) may develop and presents as tachycardia, tachypnea, exertional dyspnea, breathlessness and sweating during feeding. The child's growth is also often delayed because of poor caloric intake. In some infants, especially those with Down's syndrome, the pulmonary vascular resistance may not significantly drop. These infants may not develop CHF but are at increased risk of developing pulmonary hypertension. They may need earlier surgical intervention to prevent worsening of pulmonary hypertension and the early development of Eisenmenger syndrome.
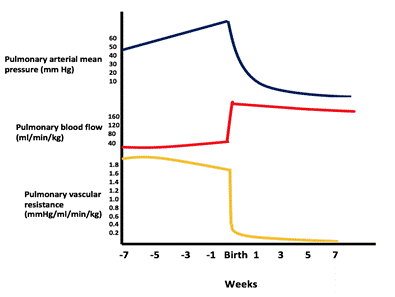
Figure showing the drop in pulmonary vascular pressure/resistance and the increase in pulmonary blood flow after birth
While more than half of small and medium sized VSDs close spontaneously, only about 10% of large VSDs close spontaneously. The muscular VSD closes by muscle in-growth. The membranous VSD closes by the neighboring tricuspid valve leaflet tissue that forms an aneurysm that gradually gets endothelialized. Both inlet and supra-cristal (outlet) VSDs are unlikely to close spontaneously. In un-operated patients with large VSD, Eisenmenger syndrome may develop within two years but may develop as early as one year in Down syndrome patients. This could be attributed to the increased vascular/alveolar density and increased secretion of endostatin in Down syndrome patients.
Asymptomatic children with a small or medium sized VSD need only supportive care, with the expectation that the VSD will close in the first few years of life. If CHF develops, treatment consists of diuretics, afterload reducing agents such as ACE inhibitors, and possibly digoxin. Heart failure in left-to-right shunts is due to volume overload to the pulmonary circulation. This is in contrast to adults with myocardial infarction in which heart failure is due to pump dysfunction. In adults with heart failure, digoxin is used to enhance pump function. However, if digoxin is used in treatment of CHF due to left-to-right shunt, it works primarily for its cholinergic effect to decrease the heart rate. Fluid restriction should be avoided as it reduces the caloric intake and delays growth.
Oxygen therapy should also be avoided as it is a pulmonary vasodilator and a systemic vasoconstrictor. That would worsen the left-to-right shunt and CHF symptoms. Patients with persistent CHF or those who are developing pulmonary hypertension require surgical closure of the VSD. If a patient is not a suitable candidate for surgery, pulmonary artery banding should be considered until surgery can be done (usually within the first year of life). Recently trans-catheter techniques have been used to close VSDs (especially the muscular VSDs).
The ductus arteriosus, formed from the embryonic 6th aortic arch, connects the aorta to the pulmonary artery. It normally closes within a few days after birth. A high oxygen tension and a decrease in endogenous prostaglandins are important factors in inducing ductal closure. For the same reason, prostaglandin synthetase inhibitors such as indomethacin are effective in inducing ductal closure and are commonly used in the neonatal period, especially in preterm infants.
Figure showing a Patent Ductus Arteriosus
Failure of closure of the ductus arteriosus leads to hemodynamic changes similar to those seen in VSD. The direction and extent of the shunt in the PDA depends on the size of the PDA and the relative systemic and pulmonary vascular resistances.
PDA is more common in females, premature infants, patients with Down Syndrome and congenital rubella syndrome. The symptoms are similar to those found in VSD and depend on the size of the shunt and direction of flow. While a small PDA is usually asymptomatic, a large PDA with significant left to right shunt may lead to CHF and eventually pulmonary hypertension.
Small and moderate sized PDA often close spontaneously especially in full term infants. PDA in premature infants may need indomethacin treatment (in the first 2-4 weeks of life) or surgical ligation. Transcatheter device closure of PDA is commonly used in older children.
Atrial septal defects involve many different parts of the atrial septum. The septum secundum defect is the most common and comprises 6-10% of all CHD. It is located in the fossa ovalis, in the location of the foramen ovale. The primum septal defect is considered a partial form of atrioventricular septal defect. The other "less common" types of ASDs are the sinus venosus and the un-roofing of the coronary sinus.
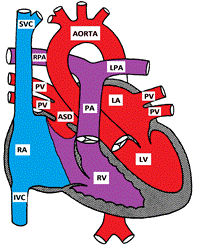
Figure showing a Secundum Atrial Septal Defect
Since the pressure difference between the two atria is small, no turbulence is generated by the flow across the ASD. In moderate to large ASDs, the direction and magnitude of the shunt across the ASD depends on the relative right and left ventricular compliances. In early infancy, the right ventricular compliance is low and the shunt across the ASD is small. As the right ventricular compliance increases, the left-to-right shunt increases. If the right ventricular compliance decreases, later in life, the shunt decreases. The shunt may eventually reverse if the patient develops pulmonary hypertension and Eisenmenger syndrome. This usually takes a few decades to develop.
ASD is usually asymptomatic and typically presents with a heart murmur in preschool age. CHF rarely develops in patients with a large ASD. Right atrial stretch may cause atrial arrhythmias. Prolonged volume overload of the lungs eventually causes pulmonary hypertension, which may take 4 to 5 decades to develop.
Most patients with ASD are asymptomatic and no specific medical management is necessary. Medical management (as in VSD) may be needed if CHF is present. Trans-catheter closure is the preferred method of closing the secundum ASDs. Surgical closure may be needed in patients with a large secundum ASD that is not amenable to trans-catheter closure or other types of ASD as they are unlikely to close spontaneously.
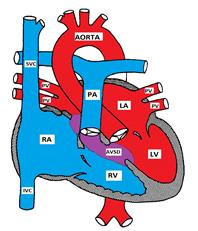
Figure Showing an Atrioventricular Septal Defect
AVSD occurs in 2% of all CHD and is more prevalent in patients with Down Syndrome. Forty percent of children with Down syndrome have CHD and 40 % of the defects are forms of AVSD
Atrioventricular septal defect is also known as endocardial cushion defect (ECD) and atrioventricular canal (AVC). It consists of a variety of defects in the endocardial cushions which form the lower part of the atrial septum, upper part (inlet) of the ventricular septum, and the medial parts of the AV valves.
There are 4 possible types:
a) Partial AVSD (primum ASD) consists of a defect in the lower part of the atrial septum and is usually associated with a cleft in the anterior mitral leaflet causing mitral insufficiency. The ventricular septum is usually intact.
b) Complete AVSD has a defect that extends from the lower part of the atrial septum to the upper part (inlet) of the ventricular septum. The mitral and tricuspid valves lose their anchor points in the ventricular septum and are instead attached to each other, forming a common AV valve that overhangs the ventricular septum.
c) Intermediate AVSD is similar to the complete AVSD but has 2 AV valves with a primum ASD and a large inlet VSD.
d) Transitional AVSD is similar to the intermediate AVSD but the VSD is small.
The pathophysiology depends on the predominant lesion (atrial vs ventricular). There is a left-to-right shunt at the atrial level due to increased relative right ventricular compliance leading to right atrial enlargement. Left atrial enlargement occurs because of mitral insufficiency secondary to the mitral valve cleft. There is a varying degree of pulmonary hypertension as the pulmonary vasculature is exposed to excess blood volume at higher pressures. Pulmonary hypertension may develop in the first two years in normal patients and in the first year in patients with Down's syndrome.
The clinical presentation of AVSD is variable and depends on the size of the defect and the degree of the left to right shunt. Patients with complete AVSD usually present with congestive heart failure in the first few weeks of life, while those with partial AVSD (primum ASD) may be completely asymptomatic.
Symptoms of CHF include poor feeding, shortness of breath, diaphoresis during feeding, and poor weight gain. Mild cyanosis may rarely develop because of right to left shunt due to increased pulmonary resistance or due to preferential streaming of the venous blood from the IVC to the left atrium.
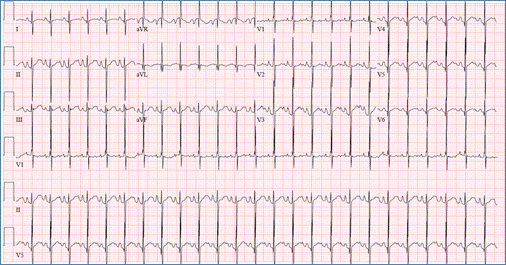
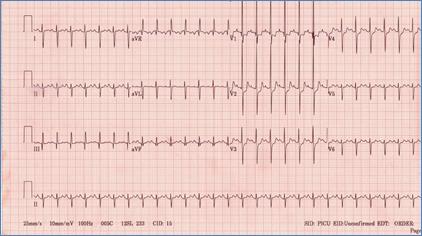
EKG showing superior QRS axis deviation (negative in avF) and right ventricular
hypertrophy in AVSD
CHF, if present, is managed with diuretics, ACE inhibitors, and occasionally digoxin. A high caloric-density formula should be used with no fluid restriction.
Almost all patients with AVSD will need surgery. The timing of surgery depends on the size of the patient, the presence of CHF, response to medical management, and the presence or absence of pulmonary hypertension. In asymptomatic patients with partial AVSD, surgery may be delayed until preschool age. Corrective surgery is usually performed in symptomatic patients with intractable CHF in the first few months of life. Even in patients who respond to medical management, the presence of Down Syndrome necessitates early surgery to prevent the development of pulmonary hypertension. If surgery cannot be performed in a symptomatic patient, pulmonary arterial banding may help limit the pulmonary blood flow until surgical repair is possible.
![]()
This section deals primarily with ventricular outflow tract obstructive lesions. Congenital mitral and tricuspid valve stenosis are relatively rare in children, thus will be not discussed here.
Outflow tract obstruction leads to an increase in the pressure proximal to the lesion and secondary myocardial hypertrophy. Turbulent flow across the obstruction produces an ejection systolic murmur. Myocardial hypertrophy leads to increased oxygen consumption with decreased exercise tolerance and possible myocardial ischemia and fibrosis. Ventricular dilatation is not a usual response to obstruction, and if present suggests ventricular failure.
Children with mild or moderate obstructive lesions usually have few symptoms. Children with severe obstruction may have low cardiac output symptoms. Newborns with severe obstruction are often PDA dependent to bypass the obstruction.
Pulmonary stenosis can be valvular, sub-valvular (infundibular), or supra-valvular.
Patients with mild or moderate PS are usually asymptomatic. Severe PS may present with decreased cardiac output symptoms such as exertional dyspnea and subsequently symptoms of congestive cardiac failure.
Balloon valvuloplasty may be done to relieve the valvular obstruction in moderate or severe cases. Surgery is indicated in supra-valvular and sub-valvular PS or if valvuloplasty is unsuccessful. Infants with critical PS need PGE1 to maintain the ductal patency before intervention.
Aortic stenosis may be valvular, supra-valvular or sub-valvular.
Aortic stenosis is more common in males. Children with mild or moderate AS are usually asymptomatic. Severe AS may be accompanied by exertional chest pain, decreased exercise tolerance or episodes of syncope. Infants with critical AS may present with CHF early in life.
Mild or moderate AS requires only supportive care. Severe valvular AS can be treated by balloon valvuloplasty. Subvalvular and supravalvular aortic stenosis are not amenable to balloon valvuloplasty and may need surgical intervention.
PGE1 infusion is essential to maintain the ductal patency in neonates with critical AS.
Surgery may involve simple valvotomy or valve replacement with a prosthesis or homograft. Another alternative is the Ross autograft procedure in which the patient's normal pulmonary valve is excised and placed in the aortic position and a homograft replaces the pulmonary valve.
Coarctation of the aorta is usually congenital, but may be acquired as in Takayasu arteritis. It may also be associated with Turner or William's syndromes, and with other CHD such as bicuspid aortic valve, VSD, or double outlet right ventricle.
Congenital coarctation is almost always juxta-ductal (opposite to the ductus arteriosus), and may be either discrete or tubular
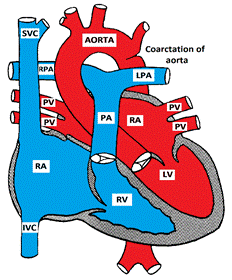
Figure showing Coarctation of the Aorta
Coarctation of the aorta causes mechanical obstruction of blood flow from the left ventricle. The pressure proximal to the coarctation is usually elevated. Collateral vessels may develop to bypass the obstruction; the most common collaterals are the internal mammary and the intercostal arteries. LVH develops in response to the elevated pressure proximal to the coarctation.
Most patients with coarctation are asymptomatic. Some patients may present with symptoms of either upper extremity hypertension (headaches, blurred vision or frequent nose bleeds) or symptoms of reduced blood flow to the lower extremities (exercise induced claudication). In infancy, severe coarctation may present with CHF as the ductus arteriosus closes. If it is not diagnosed and treated promptly, acidosis, shock and death may occur.
In asymptomatic patients without hypertension, elective surgery or angioplasty is recommended at 18 to 24 months of age. The presence of significant hypertension or CHF in infancy indicates the need for early repair. Neonates with CHF may also need medical management including PGE1 to keep the ductus arteriosus open.
![]()
In D-TGA, the aorta is positioned anterior and to the right of the pulmonary artery (instead of the normal right posterior relationship). (Figure) The aorta is connected to the right ventricle and the pulmonary artery is connected to the left ventricle.
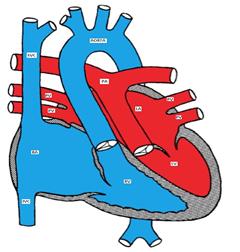
Figure showing d-transposition of the great arteries (D-TGA)
Although D-TGA is not the most common cyanotic CHD, it is the most common cardiac cause of neonatal cyanosis. It is more common in male infants of diabetic mothers who are usually large for gestational age (happy chubby blue boy).
The blood runs in two parallel circulations (rather than the normal series circulation). This situation is incompatible with life unless there is mixing between the two circulations. The mixing may occur at the atrial level (ASD or PFO), ventricular level (VSD) or at the arterial level (PDA). The atrial level communication is the most important as a bidirectional shunt can easily be produced due to the low pressure difference between the two atria as compared to the ventricles or the great arteries.
The physical examination is usually benign except for severe cyanosis and a single loud S2.
Medical management includes starting PGE1 to keep the ductus arteriosus open and treatment of metabolic acidosis if present.
Surgical repair options include (Figures):
Arterial switch (Jatene) procedure in which the great arteries are switched back to the corresponding ventricle and the coronary arteries are reimplanted in the neo-aorta.
Atrial switch (Senning or Mustard) procedure involves redirecting the blood flow at the atrial level so the pulmonary venous blood is baffled to the right ventricle (systemic ventricle) and the systemic venous blood is directed to the left ventricle (pulmonary ventricle). It is a less favorable surgery because of its numerous complications.
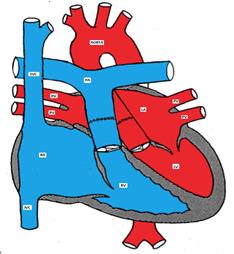
Figure showing D-TGA after arterial switch surgery.
Normal pulmonary and systemic circulations are established.
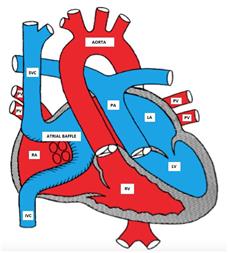
Figure showing D-TGA after atrial switch surgery
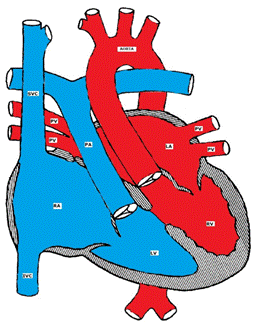
Figure showing Corrected Transposition of the Great Arteries (L-TGA)
L-TGA is a rare form of CHD (less than 1%). Both atrio-ventricular and ventricular arterial relationships are reversed, i.e. the right atrium is connected to a morphologic LV that is located on the right and the left atrium is connected to a morphologic RV located on the left (ventricular inversion). The great arteries are also transposed, i.e. the aorta is connected to the morphologic RV on the left and the pulmonary artery is connected to the morphologic LV on the right (Figure). The aortic valve is anterior and to the left of the pulmonary valve. Hemodynamically, both systemic and pulmonary circulations are in series and the patients are usually asymptomatic unless there are other associated defects. L-TGA is commonly associated with other CHD such as VSD (50%), pulmonary stenosis, left AV valve (tricuspid) insufficiency, complete AV block (AVB) and other arrhythmias.
Medical: management of CHF and arrhythmias if present. Strenuous sports restriction is indicated to prevent strain of the systemic right ventricle.
Surgical: repair of associated CHD (VSD, PS, tricuspid valve replacement). Pacemaker implantation may be needed for AVB. Double switch procedure (arterial and atrial) has been tried to treat the failing RV (Figures). Heart transplantation may be considered in some cas
Truncus arteriosus is sometimes associated with DiGeorge syndrome (deletion of chromosome 22q.11) which is characterized by hypocalcemia and reduced T-lymphocyte function. Extra-cardiac abnormalities are present in 25-40% of patients.
A single large trunk overrides a large VSD and gives rise to the coronary arteries, pulmonary arteries, and the brachiocephalic arteries (Figure).
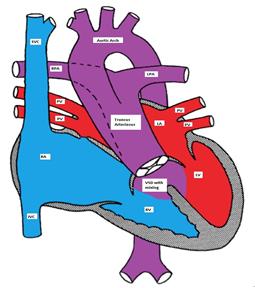
Truncus Arteriosus, with the main pulmonary artery arising from the common trunk
TA has a single ventricle physiology as the VSD is usually large (one ventricle pumps blood to both systemic and pulmonary circulations with complete mixing of blue and red blood). The truncal valve is frequently abnormal. In the neonatal period, the pulmonary vascular resistance is elevated and the pulmonary blood flow may be limited. This may cause mild and transient cyanosis due to a right-to-left shunt. As the pulmonary vascular resistance drops, the pulmonary blood flow significantly increases causing CHF symptoms.
Physical Examination: mild cyanosis may be detected along with a wide pulse pressure and cardiomegaly. A single S2 (one semilunar valve) and a systolic ejection murmur due to increased flow through the truncal valve may be heard. An apical diastolic murmur may be heard due to increased flow through the mitral valve (relative mitral stenosis). An early diastolic murmur may indicate truncal valve insufficiency.
Medical management of CHF may be tried until surgical repair can be performed.
Surgical repair may include:
Tetralogy of Fallot is the most common cyanotic CHD (over 50% of all cases of cyanotic CHD; ~ 10% of all cases of CHD).
TOF has four abnormalities (hence the term 'tetralogy):
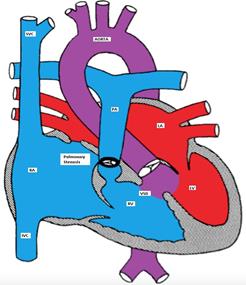
Figure showing tetralogy of Fallot (TOF) with large mal-aligned VSD, overriding aorta,
right ventricular outflow tract obstruction and right ventricular hypertrophy
The VSD is nonrestrictive (both ventricles have equal pressures). The direction of the blood flow across the VSD depends on the relative systemic and right ventricular outflow tract (RVOT) resistances.
TOF patients usually present with a heart murmur at birth with variable degree of cyanosis (depending on the degree of RVOTO). Many patients are completely acyanotic (pink TOF). There is a right ventricular tap at the left sternal border, systolic ejection murmur due to RVOTO, and single S2. The VSD is usually non-restrictive and does not produce a heart murmur. Clubbing of digits may be seen in untreated TOF with prolonged cyanosis.
|
More Information |
|
TOF with Pulmonary Atresia Pulmonary atresia may occur in 15% of patients with TOF and may be associated with hypoplasia of the peripheral pulmonary arteries. In this lesion, the pulmonary blood flow depends on the patency of the ductus arteriosus. Collaterals develop between the systemic and pulmonary circulations. The patients are usually cyanotic at birth and the degree of cyanosis depends on the degree of mixing of both circuits. The patency of the ductus arteriosus has to be maintained by PGE-1 until surgery is performed.
TOF with absent pulmonary valve This condition occurs in about 2% of patients with TOF. The pulmonary valve is usually dysplastic, regurgitant and stenotic. There is aneurysmal dilation of the proximal pulmonary arteries, which may compress the major airways causing obstruction, especially in supine position.
Clinically, the patients have respiratory distress in supine position and mild cyanosis which may disappear if CHF develops due to decrease in the pulmonary vascular resistance. To-and-fro murmur due to PS and PI is classic. Surgery is usually needed to reduce the airway compression and to repair the TOF
|
Chest X-ray showing boot shaped heart in tetralogy of Fallot
Tet spells (hypercyanotic or hypoxic spells): (hence the term 'blue baby)
Tet spells are characterized by paroxysmal cyanosis, tachycardia, tachypnea and irritability. These spells are caused by reversal of the shunt across the VSD so the blood flows from the right to the left ventricle due to an increase in the RVOTO resistance. Initially, the systolic murmur gets louder and then diminishes as the RVOTO progresses to near complete obstruction. Tet spells are more common in the warm morning hours when the systemic vascular resistance is low due to increased parasympathetic tone and decreased intravascular volume; a warm bath can have a similar effect. The RVOTO is worsened by increased catecholamine secretion due to irritability, hunger, or wet diapers.
Hypercyanotic spells need urgent treatment because if left untreated, metabolic acidosis, seizures or cerebrovascular accidents or even death may occur. The concept of treatment is to reverse the mechanism of the hypoxic spells. It is important to remove any source of irritation for the infant to decrease the catecholamine surge and worsening of the RVOTO.
While the mother is comforting the baby (nursing, bottle feeding), the baby is placed in a knee-to-chest position to increase systemic vascular resistance. Blow-by O2 is provided to enhance the oxygenation of blood and increase the systemic vascular resistance. If the spell continues, morphine sulphate (0.2 mg/kg) may be given intramuscularly to calm the patient and relax the obstruction of the RVOT. An IV line should be inserted and isotonic fluid boluses should be administered and may be repeated until the heart rate normalizes.
Acidosis should be treated with sodium bicarbonate. If the spell continues, vasoconstrictors such as phenylephrine may be tried. Intravenous beta blockers (propranolol or esmolol) may be used to decrease the cardiac contractility and hence the RVOTO. Beta blockers also slow down the heart rate and therefore enhance the coronary perfusion. If the spell continues despite all these measures, endotracheal intubation and general anesthesia are indicated in preparation for urgent surgical intervention.
If the patient is pink and has never had a cyanotic spell, elective surgery is usually performed in the first few months of life by closing the VSD and relieving the RVOTO. If the patient is cyanotic or develops a cyanotic spell, intervention is necessary in the neonatal period. If neonatal repair cannot be done because of prematurity or an unstable medical condition, palliative Blalock-Taussig shunt (anastomosis between the subclavian artery and pulmonary artery)(Figure) or its modification, a shunt between the innominate and pulmonary arteries, is performed to bypass the RVOTO.
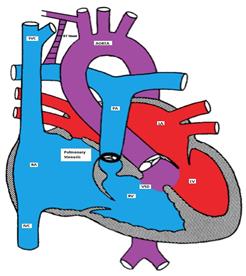
Figure showing tetralogy of Fallot (TOF) with a BT shunt between the right subclavian and the right pulmonary arteries
Pulmonary atresia may occur in 15% of patients with TOF and may be associated with hypoplasia of the peripheral pulmonary arteries. In this lesion, the pulmonary blood flow depends on the patency of the ductus arteriosus. Collaterals develop between the systemic and pulmonary circulations. The patients are usually cyanotic at birth and the degree of cyanosis depends on the degree of mixing of both circuits. The patency of the ductus arteriosus has to be maintained by PGE-1 until surgery is performed.
This condition occurs in about 2% of patients with TOF. The pulmonary valve is usually dysplastic, regurgitant and stenotic. There is aneurysmal dilation of the proximal pulmonary arteries, which may compress the major airways causing obstruction, especially in the supine position.
Clinically, the patients have respiratory distress in the supine position and mild cyanosis which may disappear if CHF develops due to a decrease in the pulmonary vascular resistance. To-and-fro murmur due to PS and PI is classic. Surgery is usually needed to reduce the airway compression and to repair the TOF.
TAPVC accounts for about 1% of all CHD. It is also referred to as total anomalous pulmonary venous return (TAPVR) which is less accurate as the connection may be obstructed and there may be diminished or no pulmonary venous return.
In TAPVC, the pulmonary veins connect to the right atrium or great veins (SVS or IVC) instead of connecting to the left atrium. An atrial communication is necessary to maintain life.
In the right atrium, both oxygenated and deoxygenated blood completely mix, and the mixture is then distributed to other cardiac chambers. The oxygen saturation is characteristically the same in all cardiac chambers. There are four different types of TVAPC:
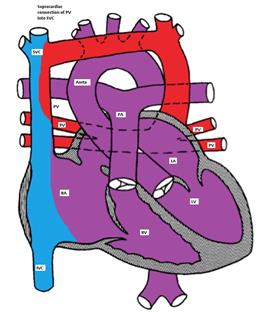
Figure showing supracardiac type TAPVC with drainage of pulmonary veins into superior vena cava (SVC) via vertical vein, which connects to the innominate vein and finally drains into SVC
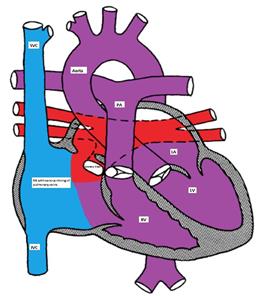
Figure showing the cardiac type of TAPVC with drainage of pulmonary veins directly into right atrium via coronary sinus
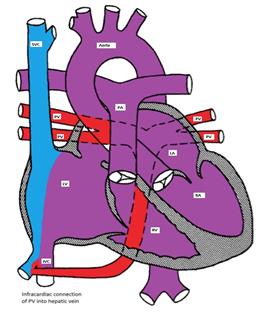
Figure showing infracardiac type TAPVC with drainage of pulmonary veins into inferior vena cava via hepatic vein
The clinical manifestations depend on the presence or absence of obstruction to the pulmonary venous connection. If there is no obstruction, mild cyanosis, CHF and frequent pulmonary infection are the common manifestations. With obstruction, there is marked cyanosis, respiratory distress and pulmonary congestion.
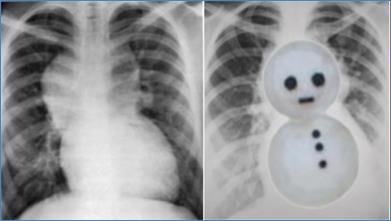
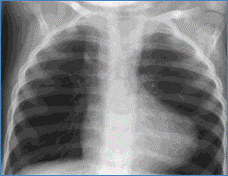
Chest X-ray showing snowman sign of supracardiac TAPVC
Medical management may be tried in patients with TAPVC without obstruction in the form of diuretics and correction of metabolic acidosis. PGE1 causes pulmonary vasodilatation and should be avoided as it may worsen the CHF. All patients with TAPVC will need surgical repair as soon as the diagnosis is made where the pulmonary veins are re-implanted in the left atrium.
TA is a rare form of CHD (1% of all CHD). The RV is hypoplastic and an ASD is necessary for survival. If the ventricular septum is intact, the pulmonary valve would also be atretic. If a VSD is present, there will be a variable degree of hypoplasia of the pulmonary valve depending on the size of the VSD.
TA is classified according to the presence or absence of VSD or TGA. In one fourth of the patients, there is a TGA. Patients with TA with normally related great vessels may be more cyanotic than those with TGA as the degree of cyanosis is inversely related to the volume of the pulmonary blood flow (Figures).
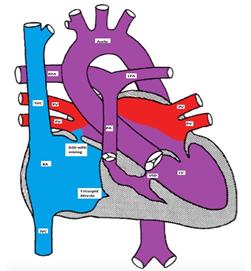
Figure showing tricuspid atresia with a restrictive VSD, pulmonary hypoplasia and normally related great arteries
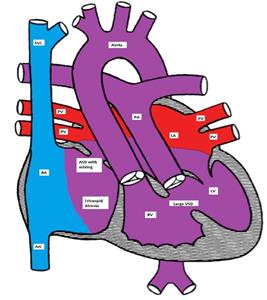
Figure showing tricuspid atresia with a non-restrictive VSD and d-TGA
Patients with TA are cyanotic and have a single S2. A holosystolic murmur of VSD may be heard. A PDA murmur may also be heard.
EKG classically shows a left superior axis deviation (due to posterior and inferior displacement of AV node) and diminished RV forces. LVH may be present. Echo is usually diagnostic.
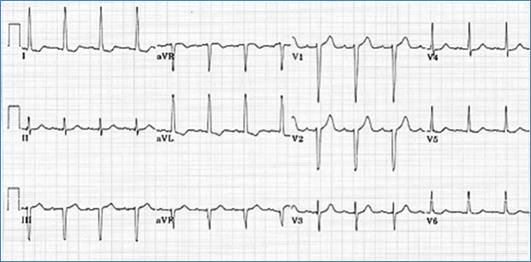
EKG showing superior left axis deviation and reduced right ventricular forces consistent with tricuspid atresia
Management includes IV infusion of PGE-1 to maintain the ductal patency. CHF needs to be treated if present. Rashkind atrial septostomy may be needed if the ASD is restrictive. Patients with TGA and hypoplastic RV will need multi-stage single ventricle repair (see Fontan-type operation in HLHS).
PA with IVS occurs in 2.5 % of patients with CHD.
There is no direct communication between the RV and the main pulmonary artery (MPA) as the pulmonary valve is atretic. The RV is usually hypoplastic. An atrial communication is needed for survival. The pulmonary blood flow is dependent on the patency of the ductus arteriosus.
Clinically, patients are severely cyanotic and have respiratory distress. The S2 is single and a PDA murmur may be heard. EKG shows RAE, LVH and possibly RVH.
CXR usually shows reduced pulmonary vasculature.
Ebstein anomaly is a rare form of CHD (1%).
There is a downward displacement of the septal and posterior leaflets of the tricuspid valve into the RV cavity creating a large atrialized portion of the RV. The functional RV may be hypoplastic, dilated and thin-walled. There is usually an inter-atrial communication such as PFO or ASD. Ebstein anomaly may be associated with other CHD such as PS, TOF, pulmonary atresia, VSD.
Dysrhythmias, especially Wolff-Parkinson-White (WPW) and SVT are commo
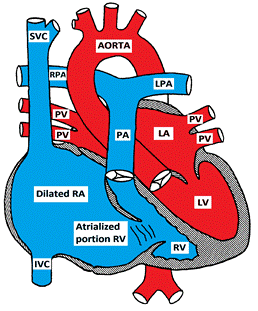
Figure showing Ebstein Anomaly of the tricuspid valve
Infants may present with cyanosis which usually improves as the pulmonary vascular resistance drops. Older children usually present with dyspnea, fatigue and palpitations (SVT). A quadruple rhythm may be audible in addition to a holosystolic murmur at the left lower sternal border due to tricuspid insufficiency. A mid-diastolic murmur due to relative tricuspid stenosis may be heard at the same location. EKG is characterized by massive right atrial enlargement (RAE), RBBB and sometimes first degree AV block or WPW. CXR shows cardiomegaly due to RAE. Echocardiogram is diagnostic. The degree of TR and the size of the RV should be determined.
Medical management is recommended in mild forms. This includes management of CHF and treatment of dysrhythmias. Oxygen supplementation is helpful early in life as it decreases the pulmonary vascular resistance. PGE-1 may be needed to maintain the ductal patency in severe cases. Surgery is required for severe forms and involves reconstruction or replacement of the tricuspid valve and closure of the ASD. If the RV is severely hypoplastic, a single ventricle repair (Fontan procedure) may be necessary.
Hypoplastic Left Heart Syndrome (HHLS) occurs in about 1% of CHD and involves hypoplasia or atresia of the left sided structures including the LA, mitral valve, LV, aortic valve and ascending aorta (Figure). A shunt at the atrial level (ASD or PFO) is necessary for survival. A PDA is essential for providing blood supply in a retrograde direction to the transverse and ascending aorta to provide blood to the neck vessels and coronary arteries.
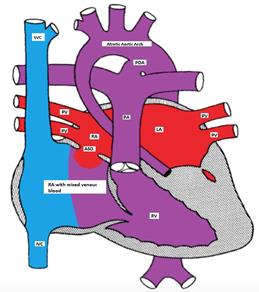
Figure showing hypoplastic left heart with a non-restrictive ASD and a PDA
There is an obligatory L to R shunt at the atrial level. Both pulmonary and systemic venous blood completely mix in the RA. The RV pumps blood to both systemic and pulmonary circulations. Initially, both resistances are nearly equal and the blood is distributed equally to the pulmonary and systemic circulations. As the pulmonary vascular resistance drops in the first few days of life, more blood flows to the lungs causing pulmonary congestion, reduced systemic flow causing tissue hypoxemia, metabolic acidosis, shock or even death.
Neonates with HLHS usually present with one of three clinical scenarios:
A systolic ejection murmur may be heard at the ULSB due to increased flow through the pulmonary valve. A holosystolic murmur due to tricuspid insufficiency may be heard at the LLSB. S2 is usually loud and single. The peripheral pulses may be weak and the skin may be mottled due to poor tissue perfusion.
Medical management:
Surgical management: both multi-stage palliative surgery and heart transplantation have comparable outcomes.
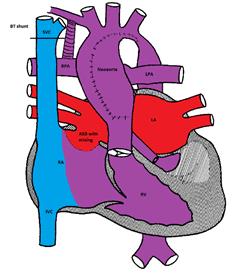
Figure demonstrating the Norwood procedure
The first stage (Norwood procedure) is performed in the neonatal period and involves division of the main pulmonary artery and ligation of the ductus arteriosus, removal of the atrial septum and reconstruction of a new ascending aorta using the proximal main pulmonary artery and the ascending aorta. A Gore-Tex® shunt (BT) is placed between the innominate artery and the right pulmonary artery to supply blood to both lungs. Because there is complete mixing of blood in the RV, the oxygen saturation is usually in the low 80's%.
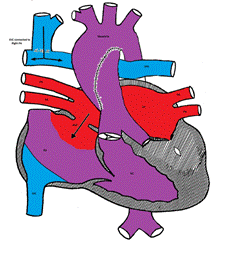
Figure demonstrating the Bidirectional Glenn procedure
The second stage is called bi-directional Glenn or hemi-Fontan procedure and is usually performed at 6 months of age. The SVC is connected to the right pulmonary artery and the BT shunt is removed. The systemic venous blood from the SVC is the only source of pulmonary circulation. The IVC venous blood mixes with the pulmonary venous blood in the RV. The systemic saturation is usually in the mid to high 80's%.
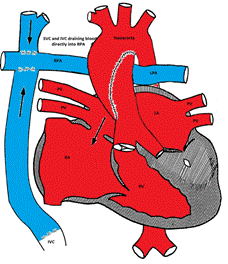
Figure demonstrating the Modified Fontan procedure
The third stage is the Fontan procedure which is usually performed at 18-24 months of age. It involves connecting the IVC to the right pulmonary artery. After this procedure, both systemic and pulmonary circulations are separated. The systemic venous blood from the SVC and IVC passively flows to both lungs via the pulmonary arteries. The RV acts as the systemic ventricle. The oxygen saturation is usually in the low to mid 90% (as there is still some mixing from the coronary venous blood via the coronary sinus).
The Fontan procedure has many complications including an increase in systemic venous pressure, development of chylothorax, and diaphragmatic and vocal cord paralysis. Other complications include atrial tachy-arrhythmias, sick sinus syndrome and protein-losing enteropathy.
The first stage (Norwood procedure) is performed in the neonatal period and involves division of the main pulmonary artery and ligation of the ductus arteriosus, removal of the atrial septum and reconstruction of a new ascending aorta using the proximal main pulmonary artery and the ascending aorta. A Gore-Tex® shunt (BT) is placed between the innominate artery and the right pulmonary artery to supply blood to both lungs. Because there is complete mixing of blood in the RV, the oxygen saturation is usually in the low 80s%.(Figure)
The second stage is called bi-directional Glenn or hemi-Fontan procedure and is usually performed at 6 months of age. The SVC is connected to the right pulmonary artery and the BT shunt is removed. The systemic venous blood from the SVC is the only source of pulmonary circulation. The IVC venous blood mixes with the pulmonary venous blood in the RV. The systemic saturation is usually in the mid to high 80s%. (Figure)
The third stage is the Fontan procedure which is usually performed at 18-24 months of age. It involves connecting the IVC to the right pulmonary artery. After this procedure, both systemic and pulmonary circulations are separated. The systemic venous blood from the SVC and IVC passively flows to both lungs via the pulmonary arteries. The RV acts as the systemic ventricle. The oxygen saturation is usually in the low to mid 90% (as there is still some mixing from the coronary venous blood via the coronary sinus). (Figure)
The Fontan procedure has many complications including an increase in systemic venous pressure, development of chylothorax, and diaphragmatic and vocal cord paralysis. Other complications include atrial tachy-arrhythmias, sick sinus syndrome and protein-losing enteropathy.
![]()
Cardiomyopathy is defined as a structural or functional abnormality of the myocardium that is not secondary to structural heart disease, hypertension, or pulmonary vascular disease. Cardiomyopathies are usually classified as follows:
This is the most common cause of cardiomyopathy in children. It may be secondary to myocarditis, coronary artery disease, and many other conditions. In familial cases, an autosomal dominant transmission is the most frequent pattern of inheritance. Autosomal recessive, X-linked and mitochondrial inheritance have also been described.
The heart in dilated cardiomyopathy is globular and is grossly dilated. The myocardium is pale and mottled and the endocardium is thin. Histologically, there is myocyte hypertrophy and degeneration and interstitial fibrosis as compared to myocarditis, in which there is myocyte necrosis and lymphocytic infiltration.
Systolic dysfunction is manifested by decreased shortening and ejection fractions which leads to increased end-diastolic volume and pressure. The blood flow to kidneys is diminished with resultant salt and water retention and worsening of CHF. Myocardial fibrosis may lead to ventricular arrhythmias secondary to the development of reentry circuits in the ventricles.
CHF signs and symptoms develop as the cardiac output decreases. These include decreased exercise tolerance and dyspnea with exertion. Infants present with poor feeding, diaphoresis during feeding, and failure to thrive. Palpitation and syncope are sometimes the earliest presentation of DCM.
If cardiac decompensation is present, intravenous inotropic agents such as dobutamine and dopamine (in renal doses) may be useful. Amrinone and milrinone also have positive inotropic effect and also reduce the afterload. When the patient is clinically stable, oral digoxin, ACE inhibitors and B-blockers (carvedilol) may be started. Diuretic therapy is useful for removing excess fluid. Patients with acute and severe decompensation may require ventricular assist devices or extracorporeal membrane oxygenation. Heart transplantation may be considered if all other measures fail.
Constrictive pericarditis occurs when a scarred, thickened, and calcified pericardium impairs cardiac filling. The pathophysiological hallmark of pericardial constriction is equalization of the end-diastolic pressures in all four cardiac chambers. This occurs because the filling is determined by the limited pericardial volume, not the compliance of the chambers themselves.
Initial ventricular filling occurs rapidly in early diastole as blood moves from the atria to the ventricles without much change in the total cardiac volume. However, once the pericardial constraining volume is reached, diastolic filling stops abruptly. The stiff pericardium also isolates the cardiac chambers from respiratory changes in intrathoracic pressures, resulting in Kussmaul's sign.
Patients with pericardial constriction typically present with manifestations of elevated systemic venous pressures and low cardiac output. Typically, there will be marked jugular venous distension, hepatic congestion, ascites, and peripheral edema. The limited cardiac output typically presents as exercise intolerance. Patients with pericardial constriction are much more likely to have left-sided or bilateral pleural effusions.
Restrictive cardiomyopathy (RCM) is a rare form of myocardial disease that is characterized by restrictive filling of the ventricles. In this disease the contractile function of the heart and wall thicknesses are usually normal, but the filling phase of the heart is abnormal. This occurs because the cardiac muscle is stiff and poorly compliant and does not allow ventricular filling. This inability to relax and fill with blood results in a back-up of blood into the atria. RCM is the least common in children, accounting for 2.5-5% of the diagnosed cardiomyopathies. The average age at diagnosis is 5 to 6 years and it appears to affect girls somewhat more often than boys. There is a family history of cardiomyopathy in approximately 30% of cases. In most cases the cause of the disease is unknown (idiopathic), although a genetic cause is suspected in most cases of pediatric RCM.
Children with RCM frequently have a history of repeated lung infections. Referral to a cardiologist occurs when symptoms of heart failure are reported or cardiomegaly is seen on a chest x-ray. In approximately 10% of cases, syncope may be the first symptom. Sudden death may also be the initial presentation in some patients.
|
Table showing the common differences between constrictive pericarditis and restrictive cardiomyopathy |
||
|
|
Constrictive pericarditis |
Restrictive cardiomyopathy |
|---|---|---|
|
History |
Previous pericarditis, cardiac surgery, trauma, radiotherapy and connective tissue disease |
May have a positive family history |
|
Extra heart sounds |
Early S3, high pitched pericardial knock. No S4 |
Later S3, low pitched triple rhythm. S4 in some cases |
|
Mitral or tricuspid regurgitation |
Absent |
Present |
|
EKG |
Normal P waves
|
Bi-atrial dilation
|
|
CXR |
Pericardial calcification in 20-30% of the cases. Normal atria |
Pericardial calcification rare. Severe atrial dilation |
|
Atrial enlargement
|
Usually mild |
Severe in most cases |
|
Equilibration of end- diastolic pressures in all cardiac chambers |
Within 5 mm Hg in nearly all cases |
Rarely occurs
|
|
MR/CT imaging |
Shows thick pericardium in most cases |
Rarely shows thick pericardium
|
|
Endomyocardial biopsy |
Normal, or non-specific abnormalities |
Shows amyloid in some cases, rarely other specific infiltrative disease |
Myocarditis is an inflammation of cardiac myocytes associated with necrosis and degeneration.
Infection with Coxsackie B virus or adenovirus is the most common cause of myocarditis. Many other viruses, as well as bacterial infections, mycoplasma, fungi, protozoa, spirochetes and rickettsia have also been reported to cause myocarditis.
The common presentation is a history of a recent viral infection followed by the development of signs and symptoms of CHF.
Acutely sick patients require intravenous inotropic agents and diuretics until they can be transitioned to oral digoxin, afterload reducing agents and diuretics. The use of anti-inflammatory and immunosuppressive therapy is still controversial.
Hypertrophic obstructive cardiomyopathy (HOCM) is the most common cause of sudden death in the young. The most important predictors of sudden death are a family history of sudden death and recurrent syncope. Mortality in children with HOCM is twice as high as adults. The condition is usually progressive and the symptoms depend on the age and mode of presentation.
HOCM is an inherited disorder of the cardiac muscle characterized by a hypertrophied, non-dilated left ventricle. The condition is associated with abnormal relaxation of the LV and sometimes with outflow tract obstruction. The mode of inheritance is most often autosomal dominant with variable penetrance and several genetic defects have been described.
The hallmark of HOCM is left ventricular hypertrophy (LVH) which is characteristically asymmetric with more involvement of the ventricular septum. This asymmetric septal hypertrophy often produces sub-aortic obstruction that is exaggerated by systolic anterior movement (SAM) of the anterior mitral valve leaflet and its apposition to the bulging septum. Histologically, there is extensive myocardial fiber disarray. The myocardial cells are disorganized and are separated by loose connective tissue. The coronary arteries sometimes run within the myocardium (intramural) and show increased intimal and medial thickening causing luminal narrowing.
Affected individuals may be asymptomatic, may have recurrent exertional dyspnea, chest pain or syncope. The condition may be recognized only after sudden death has occurred. Infants may present with CHF and the diagnosis is often missed.
Beta-blockers are helpful in the medical management by reducing the contractility and the heart rate; thereby prolonging the diastolic time. However, these agents have no effect on the degree of LV outflow obstruction or sudden death. Digoxin, other inotropic agents, and ACE inhibitors are contraindicated in HOCM as they worsen the obstruction, though they may have a role in the end-stage dilated thin-walled HOCM hearts with impaired LV systolic function. Diuretics are also usually contraindicated as they reduce the preload and enhance the LV obstruction
Surgical myomectomy may be needed to relieve the LV obstruction. Alcohol ablation of the septal perforating coronary arteries can produce a controlled infarction and relieves the sub-aortic obstruction. There is also a role for a pacemaker or implantable defibrillator (ICD).
Patients with HOCM should avoid strenuous exercise and their families should undergo genetic counseling.
Chest pain is a frequent complaint in children, with the highest incidence in the early teenage years. Musculoskeletal structures of the chest wall are the common culprits; cardiac reasons are rarely the cause but should always be considered.
A good history, including a family history, may help exclude serious causes of chest pain. Benign chest pain is not associated with other symptoms. Chest pain with exercise should raise the suspicion of potentially serious causes and warrants further investigations especially if associated with lightheadedness or presyncope. Exercise induced asthma should also be suspected.
An EKG should be done for any patient with chest pain. Further investigations including echocardiography or other imaging modalities depend on the history and physical examination. An exercise stress test may be done if the chest pain is associated with physical activities.
The management is directed to the underlying cause. Musculoskeletal chest pain and costochondritis are managed by reassurance and non-steroidal anti-inflammatory medications. Exercise induced asthma is managed with bronchodilators. Cardiac causes of chest pain are managed according to the specific etiology.
|
Summary of Normal Values |
||||||||||||||
|
Age Group |
Heart Rate (beats/min)* |
Frontal Plane QRS Vector (degrees) |
PR Interval (sec) |
QRS Duration V5 |
Q III (mm) †‡ |
Q V6 (mm) † |
RV1 (mm) |
SV1 (mm) |
R/SV1 |
RV6 (mm) |
SV6 (mm) |
R/SV 6 |
SV1+ RV 6 (mm) † |
R + S V 4 (mm) † |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Less than 1 day |
93-154 |
+59 to -163 |
0.08-0.16 (.11) |
.03-0.07 (.05) |
4.5
|
2
|
5-26 (14) |
0-23 (8) |
.1-U (2.2) |
0-11 (4) |
0-9.5 (3) |
.1-U (2.0)
|
28
|
52.5
|
|
1 to 2 days |
90-159 (123) |
+64 to -161 |
0.08-0.14 (.11) |
.03-0.07 (.05) |
6.5 |
2.5 |
5-27 (14) |
0-21 (9) |
.1-U (2.2) |
0-12 (4.5) |
0-9.5 (3) |
.1-U (2.5)
|
29 |
52 |
|
3 to 6 days |
91-166 (129) |
+77 to -163 |
0.07-0.14 (.11) |
.03-0.14 (.05) |
5.5 |
3 |
3-24 (13) |
0-17 (7) |
.1-U (2.2) |
.5-12 (5) |
0-10 (3.5) |
.1-U (2.2)
|
24.5 |
49 |
|
1 to 3 wks |
107-182 (148) |
+65 to +161 |
0.07-0.14 (.10) |
.03-0.08 (.05) |
6 |
3 |
3-12 (11) |
0-11 (4) |
.1-U (2.2) |
2.5-16.5 (7.5) |
0-10 (3.5) |
.1-U (3.3)
|
21 |
49 |
|
1 to 2 mo |
121-179 (14)9 |
+31 to +113 |
0.07-0.13 (.10) |
.03-0.08 (.05) |
7.5 |
3 |
3-8 (10) |
0-12 (5) |
.1-U (2.2) |
5-21.5 (11.5) |
0-6.5 (3) |
.2-U (4.8)
|
29 |
53.5 |
|
3 to 5 mo |
106-186 (141) |
+7 to +104 |
0.07-0.15 (.11) |
.03-0.08 (.05) |
6.5 |
3 |
3-20 (10) |
0-17 (6) |
.1-U (2.2) |
6.5-22.5 (13) |
0-10 (3) |
.2-U (6.2)
|
35 |
61.5 |
|
6 to 11 mo |
109-169 (134) |
+6 to +99 |
0.07-0.16 (.11) |
.03-0.08 (.05) |
8.5 |
3 |
1.5-20 (9.5) |
.5-18 (4) |
.1-U (2.2) |
6-22.5 (12.5) |
0-7 (2) |
.2-U (7.6)
|
37 |
53 |
|
1 to 2 yr |
89-151 (119) |
+7 to +101 |
0.08-0.15 (.11) |
.04-0.08 (.06) |
6 |
3 |
2.5-17 (9) |
.5-21 (8) |
.1-U (2.2) |
6-22.5 (13) |
0-6.5 (2) |
.3-U (9.3)
|
39 |
49.5 |
|
3 to 4 yr |
73-137 (108) |
+6 to +104 |
0.09-0.16 (.12) |
.04-0.08 (.06) |
5 |
9.5 |
1-18 (8) |
.2-21 (10 |
.1-U (2.2) |
8-24.5 (15) |
0-5 (1.5) |
.6-U (10.8)
|
42 |
53.5 |
|
5 to 7 yr |
65-133 (100) |
+11 to +143 |
0.09-0.16 (.12) |
.04-0.08 (.06) |
4 |
4.5 |
.5-14 (4) |
.3-24 (12) |
.1-U (2.2) |
8.5-26.5 (16) |
0-4 (1) |
.9-U (11.5)
|
47 |
54 |
|
8 to 11 yr |
62-130 (91) |
+9 to +114 |
0.08-0.16 (.11) |
.04-0.09 (.06) |
3 |
3 |
0-12 (5.5) |
.3-25 (12) |
.1-U (2.2) |
9-25.5 (16) |
0-4 (1) |
1.5-U (14.3)
|
45.5 |
53 |
|
12 to 15 yr |
60-119 (85) |
+11 to -130 |
0.08-0.16 (.11) |
.04-0.09 (.07) |
3 |
3 |
0-10 (4) |
.3-21 (11) |
.1-U (2.2) |
6.5-23 (14) |
0-4 (1) |
1.4-U (14.7
|
41 |
50 |
|
* 2 to 98% (mean) |
||||||||||||||
Arrhythmias are defined as disturbances in heart rate and/or conduction. Arrhythmias result from abnormal impulse formation, abnormal impulse conduction, or both. Arrhythmias may occur in children with normal hearts and/or may be associated with CHD, medications or electrolyte disturbances.
The normal range of heart rate depends on the age of the individual, ranging from 120-160 beat/min in the newborn to 60-80 beat/min in the adult. Trained athletes may normally have sinus bradycardia due to increased vagal tone. Pathological sinus bradycardia is usually secondary to an underlying condition such as hypothyroidism or medications such as beta-blockers.
Asymptomatic physiologic sinus bradycardia requires no treatment. In symptomatic bradycardia, the underlying cause should be treated and a pacemaker placement may be considered if there is no response to medical therapy.
This indicates prolongation of the PR interval more than 95th percentile for age and heart rate and is due to impairment in the AV node conduction caused by increased vagal tone, AV nodal ischemia or drugs such as digoxin and beta-blockers. It is usually reversible and does not require any treatment. First degree A-V block could be one of the cardiac manifestations of rheumatic fever (Figure).
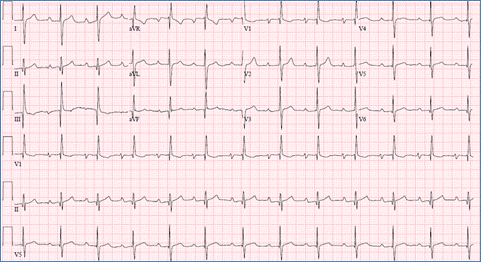
Figure demonstrating an EKG with first degree AV block
This is secondary to an intermittent failure of conduction through the AV node so that some P waves are not followed by QRS complexes.
Mobitz type I (Wenckebach) is gradual prolongation of the PR interval until there is a complete block (a P wave not followed by a QRS complex). This is due to impaired conduction through the AV node and is usually benign. This may be seen in the presence of increased vagal tone, in trained athletes and during sleep (Figure).
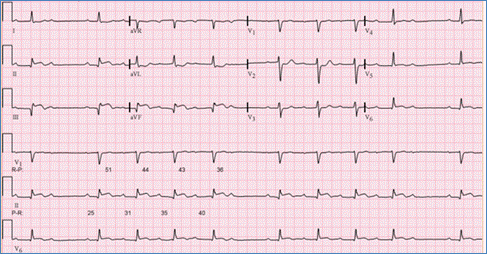
Figure demonstrating an EKG with Mobitz type I second degree AV block
Mobitz type II is sudden loss of AV conduction (two or more P waves before QRS complexes). It is more serious as it may progress to a complete AV block. Implantation of a pacemaker may be considered in symptomatic patients.
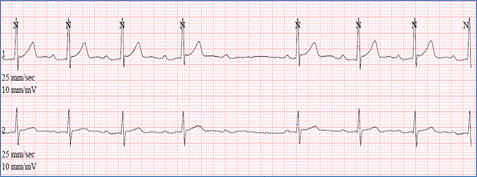
Figure demonstrating an EKG with Mobitz type II second degree AV block
Complete AV block represents complete atrioventricular dissociation with no correlation between the atrial and ventricular electrical activity. The ventricular rate is significantly slower than the atrial rate. A pacemaker placement is warranted in symptomatic patients (Figure).
This condition may be seen in infants born to mothers with systemic lupus erythematosus (SLE).
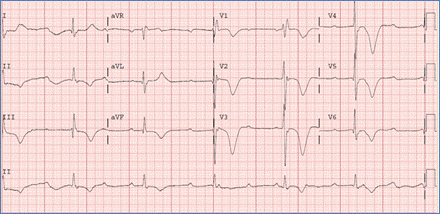
Figure demonstrating an EKG with third degree AV block
Sinus tachycardia is characterized by narrow fast QRS complexes that are preceded by normal P waves (Figure). The heart rate depends on patient age and may reach up to 220 beat/min in neonates. Sinus tachycardia may be a physiologic response to exercise, anxiety, fever, hypovolemia, hypoxemia or hyperthyroidism.
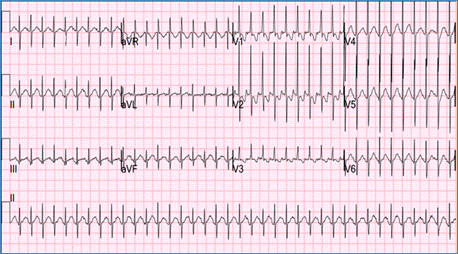
Figure demonstrating an EKG with sinus tachycardia (presence of P waves)
PACs represent origination of atrial electrical activity outside the SA node. On the EKG, PACs may appear in one of three forms:
PACs are commonly seen in infants and disappear with increasing age. This arrhythmia is usually benign and needs no treatment.
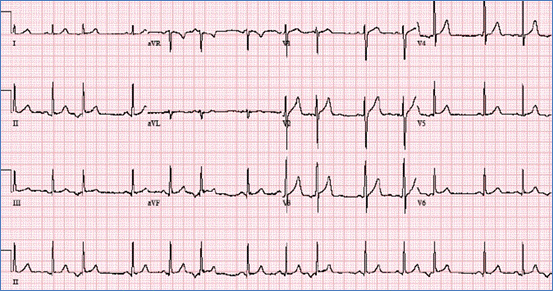
Figure demonstrating an EKG with PAC
Atrial flutter is characterized by rapid atrial activity at a rate of 200- 350 beat/min. The P waves have saw-tooth appearance. Some of the atrial impulses are not conducted through the AV node, so the ventricular rate is slower than the atrial rate and is usually regular. Atrial flutter is caused by a reentry circuit in the atrium and is usually seen in a diseased heart. The symptoms depend on the ventricular rate. Cardioversion is indicated in symptomatic patients.
Atrial fibrillation is a chaotic atrial rhythm (300-600 impulses per minute) with no well-defined P waves. The ventricular rate is irregularly irregular. Atrial fibrillation usually requires treatment with antiarrhythmic medications such as beta-blockers or Ca++ channel antagonists. Systemic anticoagulation is usually needed to reduce the risk of thrombus formation in the fibrillating atrium and downstream embolization.
SVT is characterized by a narrow QRS complex tachycardia with a heart rate of 250-350 beat/min that shows no variation with respiration (Figure). It is commonly seen in normal children but may be associated with some CHD such as Ebstein anomaly. SVT is usually caused by an accessory pathway between the atria and the ventricles, or by a reentry circuit within the AV node.
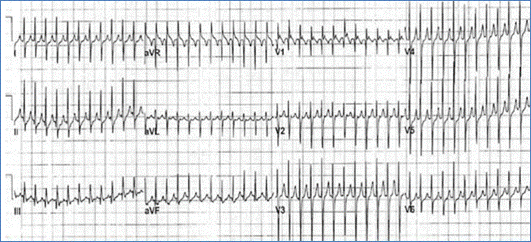
Figure demonstrating an EKG with SVT (no P waves are seen)
In infants, SVT presents with poor feeding, irritability, sweating and respiratory distress. If not treated, CHF and death may occur.
Compensated SVT should be treated promptly with vagal maneuvers such as application of ice to the face. If this is unsuccessful, then adenosine should be administered intravenously. Children with uncompensated SVT should undergo cardioversion.
WPW syndrome is an example of pre-excitation due to an accessory pathway between the atria and ventricles (Figure). It is characterized by short PR intervals, delta waves, and wide QRS complexes.
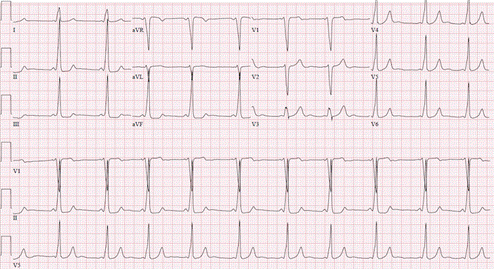
Figure demonstrating an EKG with Wolff Parkinson White syndrome
Ventricular arrhythmias are characterized by wide QRS complexes and abnormal T waves. The symptoms depend on the heart rate and are usually due to poor ventricular filling. This is a serious dysrhythmia and synchronized cardioversion is commonly indicated.
PVCs are premature, wide QRS complexes that are not preceded by P waves (Figure). Isolated unifocal PVCs originate from the same spot in the ventricles as they have uniform morphology. They are usually benign in nature and disappear with exercise. Multifocal PVCs have different morphology as they originate from different foci in the ventricles. They usually occur in diseased myocardium and often increase with exercise. If they increase with exercise, further electrophysiologic testing is usually required.
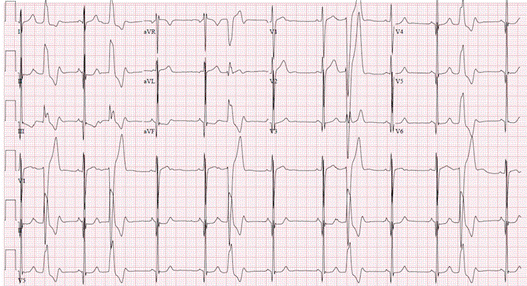
Figure demonstrating an EKG with Premature Ventricular Complexes
VT is a rapid, wide QRS-complex tachycardia with a heart rate 150-250 beat/min (Figure). It is a serious condition that may result from drug toxicity (digoxin), myocarditis or severe metabolic derangement. It should be treated promptly with cardioversion if the patient is hemodynamically unstable. Stable VT may be treated with IV lidocaine infusion. Oral amiodarone may be used for outpatient management.
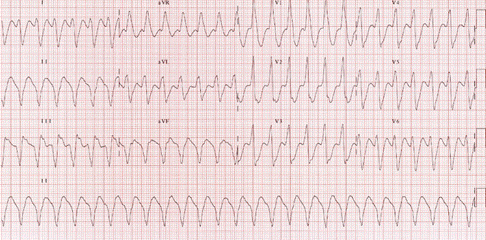
Figure demonstrating an EKG with Ventricular Tachycardia
VF is a terminal cardiac rhythm characterized by irregular wide bizarre shaped QRS complexes (Figure). It needs to be treated immediately with unsynchronized DC cardioversion.
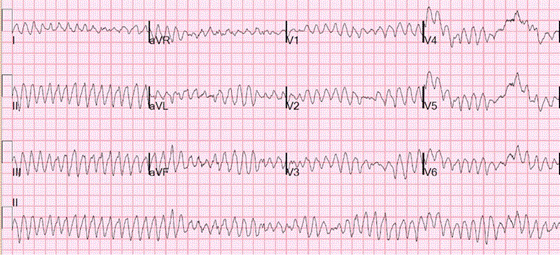
Figure demonstrating an EKG with Ventricular Fibrillation
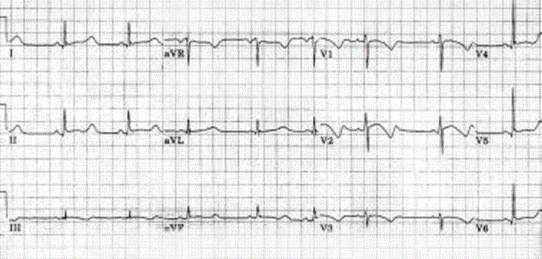
Figure demonstrating an EKG with prolonged QT interval
The QT interval represents ventricular depolarization and repolarization. The QT interval varies with heart rate. The faster the heart rate the shorter the QT interval. The corrected QT (QTc) interval is calculated by dividing the measured QT in seconds by the square root of the preceding R-R interval in seconds. A corrected QT greater than 0.46 seconds is considered prolonged (Figure). In normal neonates, the QTc may be 0.50 seconds in the first few days of life.
LQTS is an inherited condition characterized by syncope, seizures, palpitations or sudden death. There are four different classes of patients with LQTS:
|
Antipsychotics |
Antiarrhythmics |
Tricyclics |
Other antidepressants |
Antihistamines |
Others |
|---|---|---|---|---|---|
|
Chlorpromazine |
Quinidine |
Amitriptyline |
Citalopram |
Diphenhydramine |
Erythromycin |
|
Haloperidol |
Procainamide |
Doxepin |
Escitalopram |
Loratadine |
Clarithromycin |
|
Droperidol |
Disopyramide |
Imipramine |
Venlafaxine |
Terfenadine |
Quinine |
|
Quetiapine |
Flecainide |
Desipramine |
Bupropion |
|
Chloroquine |
|
Olanzapine |
Sotalol |
|
|
|
Hydroxychloroquine |
|
Thioridazine |
Amiodarone |
|
|
|
|
A table showing the common drugs which may cause QT prolongation
Congenital long QT syndrome may be caused by an imbalance of the sympathetic innervation in the heart especially the stellate ganglion or derangements in the cardiac ion flow, resulting in prolongation of the action potential. During the latter phase of the action potential, the myocardium is very excitable and may develop arrhythmia if stimulated electrically or mechanically. If a PVC occurs during this phase of the action potential (R on T phenomenon), a delayed after-depolarization develops in the form of a specific polymorphic ventricular arrhythmia (Torsades de pointes) (Figure).
Pathophysiology
Congenital long QT syndrome may be caused by an imbalance of the sympathetic innervation in the heart especially the stellate ganglion or derangements in the cardiac ion flow, resulting in prolongation of the action potential. During the latter phase of the action potential, the myocardium is very excitable and may develop arrhythmia if stimulated electrically or mechanically. If a PVC occurs during this phase of the action potential (R on T phenomenon), a delayed after-depolarization develops in the form of a specific polymorphic ventricular arrhythmia (Torsades de pointes) (Figure).
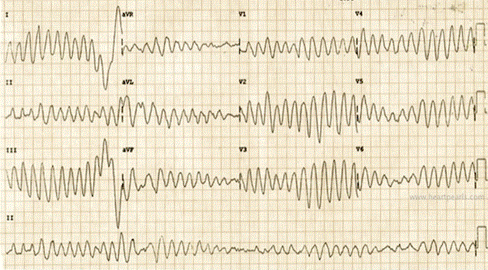
EKG showing Torsades de pointes
Most patients are usually asymptomatic. Symptomatic patients present with episodic dizziness, palpitations, syncope, seizures, and/or cardiac arrest. Exercise, emotion and loud noises are triggers for prolonged QT conversion to other arrhythmias. Prolonged QT can convert to Torsades de pointes, bradyarrhythmias, or AV block.
LQTS is a serious condition and the risk factors for sudden death include long QT > 0.55 seconds, family history of sudden death, bradycardia for age, and a prior history of symptoms. Any medications that may cause QT prolongation should be discontinued.
Treatment is aimed at preventing conversion to other possibly fatal arrhythmias. Beta-blockers are the mainstays of medical management. An implanted cardioverter-defibrillator (ICD) is often used as non-pharmacological treatment. Other non-pharmacological treatments include permanent dual chamber pacemaker and left cardiac sympathetic denervation.
![]()
Kawasaki Disease (KD) is an acute multi-system immune-mediated vasculitis of unknown etiology. It usually presents in infancy and early childhood with 85% of those affected are less than 5 years of age. KD is the leading cause of acquired heart disease in children in the US. In untreated KD patients, the incidence of coronary artery aneurysm ranges from 15-25%. Myocardial infarction from thrombotic occlusion of the coronary arteries is the principle cause of death. KD is more common in patients of Asian descent. The prognosis depends on the extent of the cardiac involvement and the promptness of medical treatment.
The etiology of KD is unknown. The inflammation in KD involves small to medium-sized arteries, including the coronary arteries. The extent of the coronary artery involvement is the critical factor that determines morbidity and mortality. Investigators propose that mediators such as tumor necrosis factor (TNF), interleukin (IL)-1B, interferon (INF) and IL-6 produced by activated T-cells and macrophages promote vascular injury. The earliest pathological change reported in the vessel wall is subendothelial accumulation of T-cells, mononuclear cells, macrophages and monocytes.
KD is divided into three phases.
According to U.S. and Japanese guidelines, Kawasaki disease is a clinical diagnosis.
In some cases, patients do not fulfill the classic criteria for Kawasaki disease and are classified as having incomplete (atypical) disease. Incomplete disease is more common in younger infants and older children and should be suspected when patients have a fever for at least five days with only two or three of the principal clinical features. As a result, it is important to consider the diagnosis of Kawasaki disease and the possible need for echocardiography in all children who have an unexplained fever lasting at least seven days with laboratory evidence of systemic inflammation .
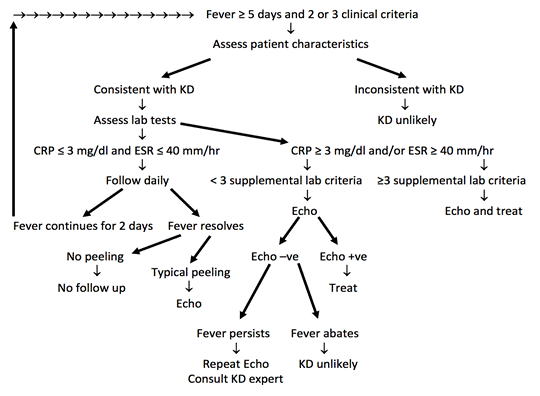
Figure demonstrating a suggested algorithm for the management of suspected Kawasaki Disease
Early detection of KD and prompt treatment reduce mortality below 1%. Baseline echocardiogram should be performed as soon as the diagnosis is made. The table below helps to stratify the risk of patients with KD and helps determine follow up testing.
1.Treatment of Acute Disease:
2. Treatment of Refractory Disease:
3. Follow up of KD
|
Risk Level |
Pharmacological Therapy |
Physical Activity |
Follow-up and Diagnostic Testing |
Invasive Testing |
|---|---|---|---|---|
|
I (No coronary artery changes at any stage of illness) |
None beyond 1st 6-8 weeks |
No restrictions beyond 1st 6-8 weeks |
Cardiovascular risk assessment, counseling at 5-y intervals |
None recommended |
|
II (transient coronary artery ectasia, disappears within 1st 6-8 weeks) |
None beyond 1st 6-8 weeks |
No restrictions beyond 1st 6-8 weeks |
Cardiovascular risk assessment, counseling at 3- to 5-y intervals |
None recommended |
|
III (1 small-medium coronary artery aneurysm / major coronary artery) |
Low-dose aspirin (3-5 mg/kg aspirin per day), at least until aneurysm regression is documented |
For patients <11 y old, no restriction beyond 1st 6-8 weeks: patients 11-20 y old, physical activity guided by biennial stress test, evaluation of myocardial perfusion scan; contact or high-impact sports discouraged for patients taking antiplatelet agents
|
Annual cardiology follow up with echocardiogram + EKG, combined with cardiovascular risk assessment, counseling; biennial stress test/evaluation of myocardial perfusion scan |
Angiography, if noninvasive test suggests ischemia |
|
IV (≥ 1 large or giant coronary artery aneurysm, or multiple or complex aneurysms in same coronary artery, without obstruction |
Long-term antiplatelet therapy and warfarin (target INR 2.0-2.5) or low-molecular-weight heparin (target: anti-factor Xa level 0.5-1.0 U /mL) should be combined in giant aneurysms |
Contact or high-impact sports should be avoided because of risk of bleeding; other physical activity recommendations guided by stress test/evaluation of myocardial perfusion scan outcome |
Biannual follow-up with echocardiogram + ECG; annual stress test/evaluation of myocardial perfusion scan |
1st angiography at 6-12 mo or sooner if clinically indicated; repeated angiography if noninvasive test, clinical or laboratory findings suggest ischemia; elective repeat angiography under some circumstances (see text)
|
|
V (coronary artery obstruction) |
Long-term low-dose aspirin; warfarin or low-molecular-weight heparin if giant aneurysm persists; consider use of β-blockers to reduce myocardial o2 consumption |
Contact or high-impact sports should be avoided because of risk of bleeding; other physical activity recommendations guided by stress test/myocardial perfusion scan outcome |
Biannual follow-up with echocardiogram and ECG; annual stress test/evaluation of myocardial perfusion scan |
Angiography recommended to address therapeutic options |
American Academy of Pediatrics.
Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: A Statement for Health Professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics 114 (6) December 2004, pp. 1708-1733 (doi:10.1542/peds.2004-2182)
Sanchez-Manubens J, Bou R, Anton J. Diagnosis and classification of Kawasaki Disease. Journal of Autoimmunity. 48-49 (2014) 113-117
Infective endocarditis (IE) is an inflammation of the endothelial lining of the heart muscle, valves and great vessels. The valves have a particularly high propensity for infection due to the lack of blood supply and limited access to immune cells. IE is relatively rare in children. It has an estimated annual incidence of 3 to 9 cases per 100,000 persons in industrialized countries. The highest rates are observed among patients with prosthetic valves, intracardiac devices, unrepaired cyanotic congenital heart diseases, or a history of infective endocarditis. About 50% of cases of infective endocarditis develop in patients with no known history of valve disease. Other risk factors include chronic rheumatic heart disease (which now accounts for <10% of cases in industrialized countries), age-related degenerative valvular lesions, hemodialysis, and coexisting conditions such as diabetes, human immunodeficiency viral infection, and intravenous drug use.
Bacteremia and the presence of endothelial damage are important factors in the pathogenesis of IE. Cyanosis and polycythemia, if present, increase the viscosity of the blood and further enhance the likelihood of developing IE. Foreign materials such as prosthetic valves or shunts also significantly increase the risk for developing IE. It is not surprising that cyanotic CHD with an artificial shunt or prosthetic valves constitutes the highest risk for IE.
Neonatal endocarditis frequently occurs on the right side of the heart and is associated with disruption of endocardium or valvular endothelial tissue by catheter-induced trauma in hospitalized infants. Premature neonates often experience transient episodes of bacteremia from trauma to the skin and mucous membranes, vigorous endotracheal suctioning, parenteral hyperalimentation, or placement of umbilical or peripheral venous catheters. The combination of endothelial damage and bacteremia is critical for the induction of IE.
Vegetations develop at the site of endothelial damage, which is usually located at the lower pressure side of the lesion i.e. in the right ventricle in patients with VSD and on the atrial surface of the mitral valve with mitral insufficiency.
After bacteria adhere to the damaged endothelium, platelets and fibrin are deposited over the organisms, leading to the formation of a vegetation. The organisms trapped within the vegetation are protected from phagocytic cells and other host defense mechanisms.
Viridans group streptococcus, enterococci and S. aureus are responsible for most cases of IE. Streptococcus pneumoniae, coagulase negative staphylococcus, gram negative bacilli and fungi may also cause IE. The type of pathogens depends on the following factors: i) whether the valve is a native or a prosthetic valve, ii) patient age and iii) source of infection.
The blood cultures may be negative in patients who have already received antibiotics or in patients who have IE caused by fastidious microorganisms such as petronella species, brucella species, Coxiella burnetii, bacteria in the HACEK group (haemophilus species, actinomycetemcomitans, Cardiobacterium hominis, Eikenella corrodens and Kingella kingae), and Tropheryma whipplei. In these cases, serologic testing, blood polymerase chain reaction (PCR) assay and highly specialized microbiologic techniques may lead to the identification of the pathogen in up to 60% of cases.
Persistent or recurrent low grade fever is the most common symptom of IE. Other symptoms are nonspecific and include malaise, myalgia, arthralgia, anorexia, night sweats and headaches. Splenomegaly can be found in 15-50% of patients with IE. A new or changing murmur indicates valvular involvement.
The classic peripheral manifestations of IE are rarely seen nowadays. These include petechiae, splinter hemorrhages (hemorrhages in the nail beds), Osler nodules (small, tender nodules on the pads of fingers and toes), Janeway lesions (painless hemorrhages on palms and soles), and Roth spots (hemorrhages in the retina with a white center).
The Duke Criteria for diagnosis of infective endocarditis: Requires 2 major + 1 minor OR 1 major + 3 minor OR 5 minor criteria for diagnosis
| Major criteria |
Minor criteria |
|---|---|
|
Positive blood Cx for IE |
Predisposing heart condition or IV drug use |
|
Typical micro-organism for IE from 2 separate blood Cx 1 |
Fever > 38° C |
|
Evidence of endocardial involvement2 |
Vascular phenomena (arterial emboli, septic pulmonary infarcts, conjunctival hemorrhage, intracranial hemorrhage and Janeway lesions) |
|
|
Immunologic phenomena (glomerulonephritis, Osler nodes, Roth spots, rheumatoid factor) |
|
|
Positive blood Cx but not meeting major criteria |
|
|
Echocardiographic findings consistent with IE but not meeting major criteria |
* : Viridans streptococci, Streptococcus bovis and HACEK group OR Staph aureus/enterococci in the absence of a primary focus and persistently positive blood Cx defined as 2 blood Cx drawn 12 hours apart
# : Positive echocardiogram for vegetations OR new valvular regurgitation
The management of IE includes 4-6 weeks of high dose IV antibiotics. The choice of antibiotics depends on the organism isolated and the results of antibiotic sensitivity testing. Initial empiric therapy may be based on the table below. Surgical intervention may be necessary if vegetations are causing obstruction, or if there is significant malfunction of a prosthetic valve.
|
Antibiotic |
Dosage |
|---|---|
|
Native valves or prosthetic valves 1 year after surgery |
|
|
Ampicillin + Oxacillin + Gentamicin |
12 g/day IV in 4-6 doses
12 g/day IV in 4-6 doses
3 mg/kg/day IV in 1 dose |
|
Prosthetic valves within 1 year of surgery |
|
|
Vancomycin + Gentamicin + Rifampin |
30 mg/kg/day IV in 2 doses
3 mg/kg/day IV in 1 dose
900-1200 mg IV/PO in 2 or 3 divided doses |
Antimicrobial prophylaxis is indicated in patients undergoing dental procedures who have:
a) Unrepaired cyanotic CHD including those with palliative shunts and conduits,
b) Completely repaired CHD with a prosthetic material or device during the first 6 months after the procedure
c) Repaired CHD with a residual defect
Antibiotics are NOT recommended for patients who have procedures involving the reproductive, urinary or gastrointestinal tract.
Prevention of Infective Endocarditis. 2007 Guidelines From the American Heart Association. Circulation. 116:1736-1754.
Ferrieri, P, et al. Unique Features of Infective Endocarditis in Childhood. Circulation. 2002;105:2115. Scientific statement from American Heart Association
Hoen B , Duval X . Clinical practice. Infective endocarditis. N Engl J Med . 2013 Apr 11;368(15):1425-33
Acute rheumatic fever (ARF) is an inflammation of the heart, skin, joints and/or brain which develops after infection with Group A streptococci, such as "strep" throat, or scarlet fever. Although the incidence of ARF has declined in Europe and North America over the past 4 to 6 decades, the disease remains one of the most important causes of cardiovascular morbidity and mortality in the developing countries that are home to the majority of the world's population. There is a 2-3 weeks delay between the strep infection and the development of ARF. Less than 3% of those with untreated strep infection may develop ARF. Most of the affected patients are between six and fifteen years of age.
Group A streptococcal (GAS) infection of the pharynx is usually the precipitating cause of rheumatic fever. During epidemics over a half a century ago, as many as 3% of untreated acute streptococcal sore throats were followed by rheumatic fever; in endemic infections, the incidence of rheumatic fever is substantially less. Appropriate antibiotic treatment of streptococcal pharyngitis prevents acute rheumatic fever in most cases. Unfortunately, at least one third of episodes of acute rheumatic fever result from inapparent streptococcal infections. In addition, some symptomatic patients may not seek appropriate medical care. Among the most widely accepted concepts in support of an autoimmune hypothesis involving the GAS was the observation by Stollerman. He noted a correlation between outbreaks of GAS upper respiratory tract infections associated with a relatively limited number of M-protein types which were followed by outbreaks of rheumatic fever. Subsequent reports demonstrated that only a limited number of specific M-types (M-5, M-6, M-18) were isolated during rheumatic fever outbreaks. These findings supported the concept of antigenic mimicry. In ARF, antibodies against the M antigen found in the streptococcal cell wall cross react with cardiac myosin causing carditis.
|
Features suggestive of GAS infection |
Features suggestive of viral infection |
|---|---|
|
Sudden onset sore throat with painful swallowing |
Conjunctivitis |
|
Fever |
Coryza |
|
Scarlet fever rash |
Hoarseness |
|
Headache, nausea and vomiting |
Cough |
|
Tonsillar exudates with soft palate petechiae |
Diarrhea |
|
Tender enlarged anterior cervical nodes |
Characteristic exanthems |
|
Patient age 5-15 years with a h/o exposure |
Characteristic enanthems |
Prevention of rheumatic fever requires adequate therapy for GAS pharyngitis. In selecting a regimen for the treatment of GAS pharyngitis, physicians should consider various factors, including bacteriologic and clinical efficacy, ease of adherence to the recommended regimen (frequency of daily administration, duration of therapy, and palatability), cost, spectrum of activity of the selected agent, and the potential side effects.
|
Agent |
Dose |
Duration |
|---|---|---|
|
Penicillins |
|
|
|
Penicillin V |
< 27 kg: 250 mg BID/TID
>27 kg: 500 mg BID/TID |
10 days
10 days |
|
Amoxicillin |
50 mg/kg daily |
10 days |
|
Benzathine Penicillin G |
<27 kg: 600,000 U IM
>27 kg: 1.2 million U IM |
Once
Once |
|
Penicillin allergic |
|
|
|
Cephalexin/Cefadroxil |
Variable |
10 days |
|
Azithromycin |
12 mg/kg (max 500 mg) daily |
5 days |
|
Clindamycin |
20 mg/kg divided in 3 doses (max 1.8 g/day) |
10 days |
Signs and symptoms of rheumatic fever include fever, migratory arthritis in large joints, abdominal pain, erythema marginatum (a ring-shaped rash located on trunk and upper parts of arms and legs), Sydenham chorea, subcutaneous nodules, epistaxis, shortness of breath and chest pain.
According to Jones Criteria, there must be evidence of previous streptococcal infection.
Any of the following may serve as evidence of GAS infection:
For ARF diagnosis, 2 major OR one major + two minor criteria are needed.
Patients with a documented history of ARF should receive antibiotic prophylaxis until the age of 21 or for a minimum of five years if there is no cardiac involvement. Patients with valvular abnormalities should receive lifetime prophylaxis. Prophylaxis consists of monthly injections of benzathine penicillin; alternatively, twice daily oral penicillin V may be used. Oral sulfadiazine may be used for patients with penicillin allergy, but it is not as effective as penicillin.
Prevention of Rheumatic Fever and Diagnosis and Treatment of Acute Streptococcal Pharyngitis: A Scientific Statement From the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee of the Council on Cardiovascular Disease in the Young, the Interdisciplinary Council on Functional Genomics and Translational Biology, and the Interdisciplinary Council on Quality of Care and Outcomes Research: Endorsed by the American Academy of Pediatrics, 2004.
Bach DS. Revised Jones Criteria for Acute Rheumatic Fever ACC, May 2015
![]()