



Disorders of the endocrine part of pancreas
Glucose is an important source for brain energy metabolism and extensive regulatory mechanisms are in place to ensure protection from hypoglycemia. Glucose concentrations naturally reach a nadir a couple hours after birth and then begin to rise reaching normal values by day 3 of life. This is related to the abrupt cessation of placental glucose transfer at delivery causing a transient decrease in glucose levels with subsequent response of increased glucagon, decreased insulin levels, and an increase in catecholamine secretion to gradually normalize plasma glucose concentration. Regulatory mechanisms in older children are balanced by gluconeogenesis and glycogenolysis. Hypoglycemia definition varies based on the age group (40 mg/dL or below in neonates) and < 55-60 mg/dL in older children.
Glucose Homeostasis
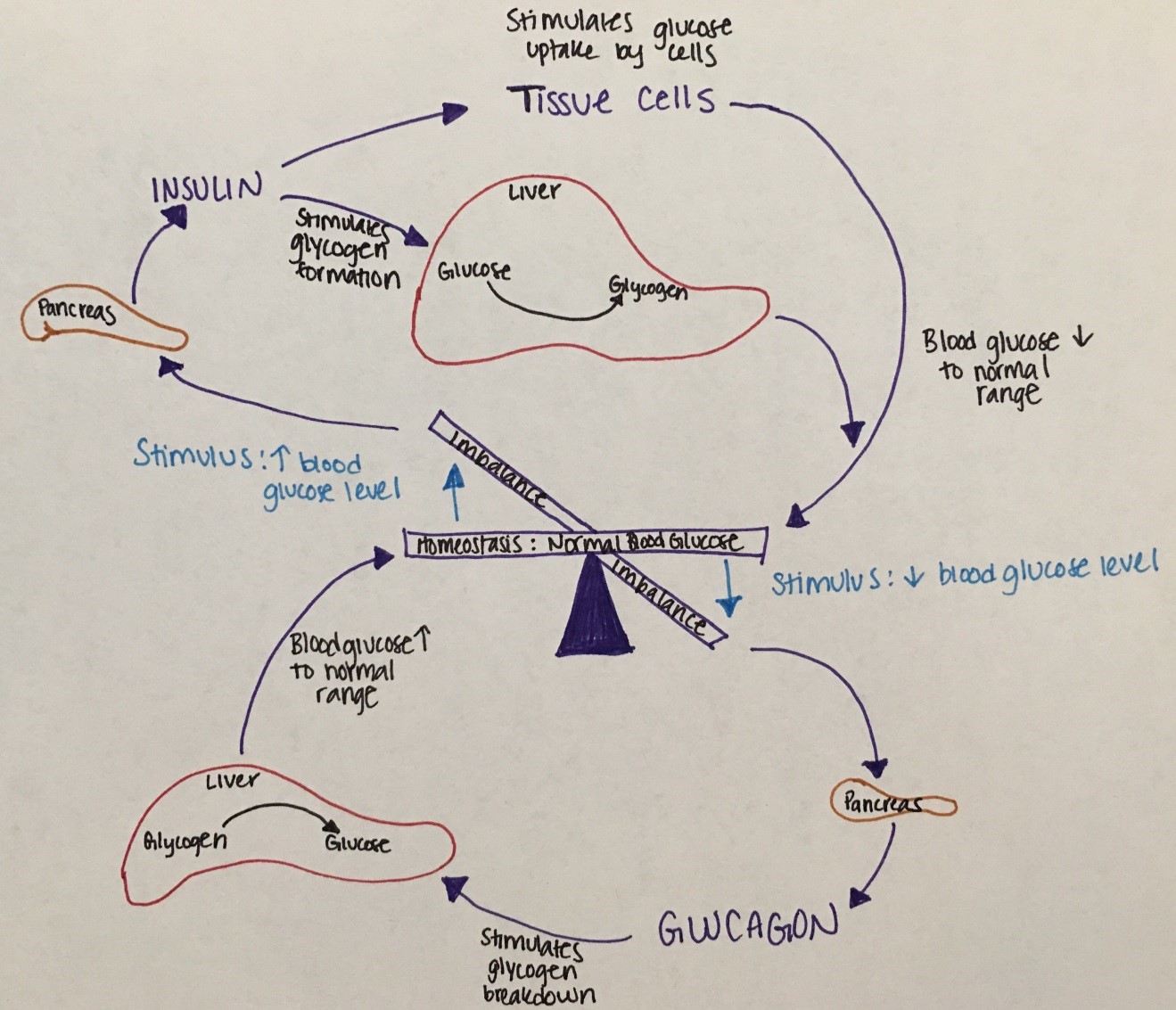
Figure 16. This figure describes the balance of glucose regulatory mechanisms.
Lower blood glucose levels stimulate the pancreas to release glucagon to stimulate glycogen breakdown in the liver to increase blood glucose levels.
High blood glucose levels stimulate insulin release from the pancreas to stimulate glycogen formation in the liver to lower blood glucose levels.
Hypoglycemia
Etiology: Hypoglycemia is due to defects of the hormones or enzymes of the glucose regulatory mechanisms that result in inadequate glucose or surplus of insulin.
|
Age |
Etiologies |
|---|---|
|
Infant |
|
|
Toddler |
|
|
Child |
|
Table 9. Etiologies of Hypoglycemia by Age
Symptoms: An abrupt decrease in plasma glucose causes adrenergic symptoms (due to catecholamine release) such as pallor, sweating, tachycardia, tremor, and emesis. A slow decrease in plasma glucose causes neuroglycopenic symptoms such as confusion, diplopia, headache, dizziness, lethargy, seizure, and lack of coordination. Neonatal symptoms of hypoglycemia are more subtle, such as apnea, low temperature, poor tone, jitteriness, and poor feeding.
Diagnosis: Critical sample needs to be obtained when BG at or <40 mg/dL and includes serum free fatty acids, ketones, and insulin levels. Hormones to be also obtained include cortisol, ACTH, and growth hormone. Acylcarnitine and total carnitine, urine organic acids and serum amino acids to be considered based on the age group/ index of suspicion. If a child has hepatomegaly with hypoglycemia, suspect an enzyme deficiency. Males with a microphallus and hypoglycemia should be evaluated for hypopituitarism. Accidental ingestion of alcohol or salicylates can cause hypoglycemia so toxicology screen and history taking will help with diagnosis in this instance. Finally, if Munchausen by Proxy is suspected, then c-peptide along with insulin levels need to be obtained to investigate for exogenous insulin administration.
Treatment: Acute treatment includes early feeding (neonates), dextrose gel, or IV boluses of 2 mL/kg of 10% dextrose in water followed by continuous infusions of glucose. If hypoglycemia persists, it is then treated with continuous glucose infusion, diazoxide and/or somatostatin analogs. If hypoglycemia persists, pancreatectomy may be necessary.
Diabetes Mellitus
Diabetes mellitus (DM) is a common metabolic condition of hyperglycemia caused by complete or partial insulin deficiency and its actions.
Insulin physiology: Insulin is made on the ribosomes of pancreatic islet beta cells as a proinsulin precursor (single chain: chain A is connected to chain B by c-peptide) that is then cleaved into insulin and c-peptide molecule. It is then released into circulation as a double chain polypeptide linked by disulfide bridges. C-peptide has a longer half-life and later nadir.
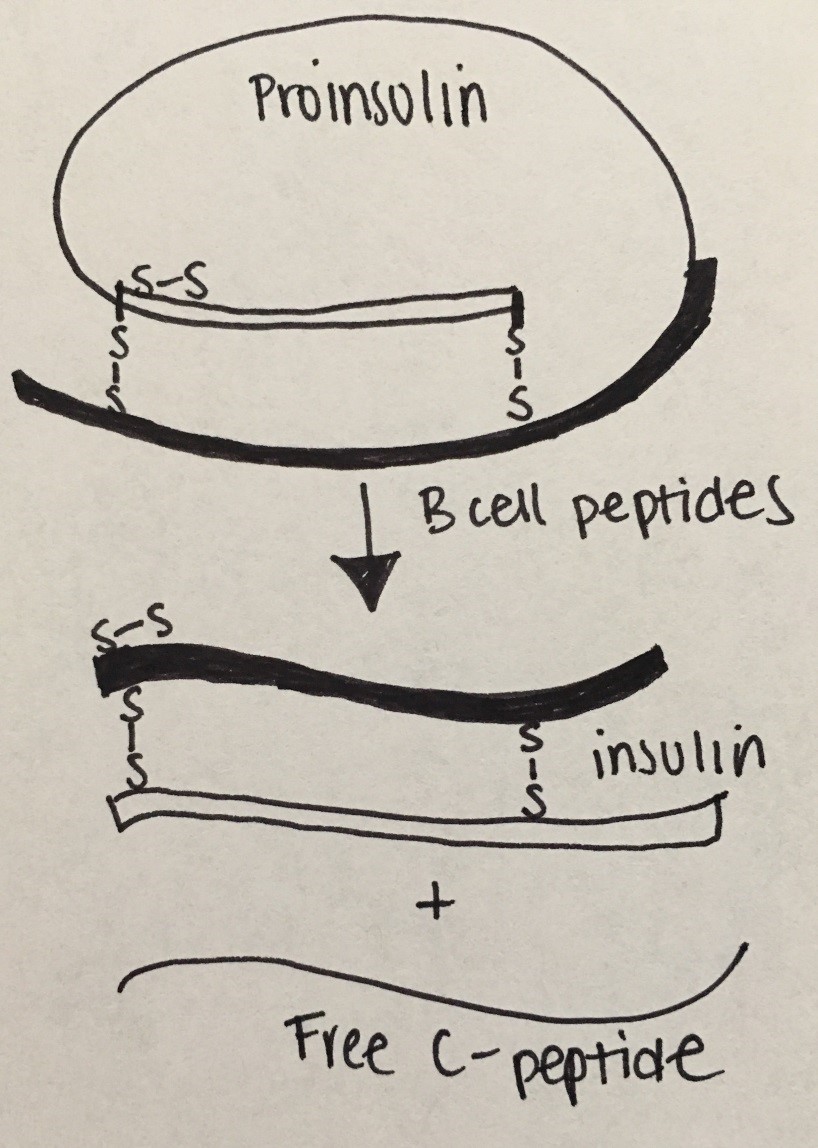
Figure 17: Insulin Physiology
Etiologies: There are many subgroups with distinct etiologies but the most commonly known will be discussed.
Type 1 DM (previously referred to as Juvenile Onset Diabetes): T1DM is due to autoimmune destruction of pancreatic beta cells resulting in insulin deficiency. Histocompatibility locus antigens that determine response to self or other antigens create antibodies to cytoplasmic and cell surface components of the islets cells, to insulin, to GAD (glutamic acid decarboxylase or enzyme in pancreatic islet innervation), and to IA-2 (islet phosphatase). T1DM is the most common type in childhood and adolescence (7-15 year olds); but with the epidemic increases in childhood obesity, there is an increased prevalence in T2DM among this age. Wasting and diabetic ketoacidosis are complications from undiagnosed/ untreated hyperglycemia.
Type 2 DM: T2DM is typically due to insulin resistance with some B-cell impairment. The insulin resistance can be observed at the level of skeletal muscle, liver, and adipose tissue, but can result in actual insulin deficiency. For more info regarding the differences between T1DM and T2DM see table below:
|
|
Type 1 |
Type 2 |
|---|---|---|
|
Insulin Deficiency: |
Absolute |
Relative |
|
Age of Onset |
75% < 21 yrs.
|
75% > 40 yrs |
|
Weight |
Thin |
Overweight |
|
Edtiology: |
Autoimmune |
Sedentary lifestyle |
|
Inheritance: |
HLA-Assn |
Complex |
|
Treatment: |
Insulin |
Healthy nutrition, excercise, oral agents |
Table 10. Comparison of Type 1 and Type 2 Diabetes Mellitus.
Monogenic diabetes (MODY or maturity onset of diabetes of youth). MODY has a dominant inheritance pattern with the mutations of the genes encoding enzymes or transcription factors affecting islet cell development and insulin secretion. This causes GLUT2 transporter regulation to be mutated.
Symptoms: polyuria, polydipsia, weight loss (T1DM); obesity (T2DM) and acanthosis nigricans. Complications: retinopathy, nephropathy, neuropathy, ischemic heart disease, vasculopathy, diabetic ketoacidosis, nonketotic hyperosmolar coma.
Diagnosis: Initial laboratory workup includes random BG or fasting BG, HbA1C, UA. BMP and blood gas are typically obtained if there is concern for DKA. Celiac panel, thyroid function tests, c-peptide and autoimmune diabetes antibodies studies (i.e. anti-GAD, anti-islet, etc) are obtained if T1DM is suspected.
Laboratory findings between T1DM and T2DM
|
|
T1DM |
T2DM |
|---|---|---|
|
Insulin |
Low |
Normal or Elevated |
|
Random Glucose |
> 200 |
>126 |
|
Background |
Autoimmune |
Genetic basis |
|
Ketosis |
Yes |
No |
|
Associated Diseases |
APS-1, Addison's Disease |
Metabolic Syndrome |
Table 11. Laboratory findings for T1DM and T2DM
|
|
Normal |
Mild |
Moderate |
Severe |
|---|---|---|---|---|
|
CO2 |
20-28 |
16-20 |
10-15 |
<10 |
|
pH |
7.35-7.45 |
7.25-7.35 |
7.15-7.25 |
<7.15 |
|
Clinical |
No change |
Oriented but fatigued |
Kussmaul respirations, oriented but sleepy |
Kussmual or depressed respirations; sleep to depressed sensorium or coma |
Table 12. Classification of DKA (from Nelson Textbook of Pediatrics)
Treatment:
T1DM: Patients are typically dependent on exogenous insulin; fluids; diet.
T2DM: First line treatment entails a diabetic diet (healthy, sugar avoidance, and CHO restriction) coupled with increased physical exercise. Oral hypoglycemic are typically introduced at the time of diagnosis. Exogenous insulin treatment is reserved for severe forms that have had DKA or HgA1C > 9%.

