



Ambiguous genitalia and disorders of sexual development (DSD)
Sex determination and sex differentiation can be divided into a) chromosomal sex or karyotype (46 XX, 46 XY, or variants), b) gonadal sex (presence of a testis or ovary) and c) phenotypic sex (external genitalia and internal structures).
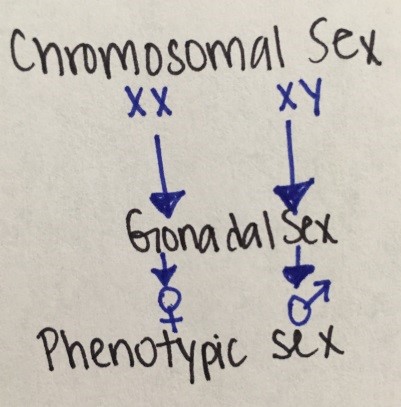
Figure 12. Sexual development
Human embryo is bipotential with genetic sex determined at conception. Male differentiation requires specific molecular and biochemical stimuli at specific times. Female differentiation occurs in absence of male determinant genes.
Disorders of sexual development can be associated with ambiguous genitalia (i.e., external genitals neither appearing clearly male nor female).
|
Sex Chromosome DSD |
46, XY DSD |
46, XX DSD |
|---|---|---|
|
45, X (Turner Syndrome & variants) |
Disorders of testicular development |
Disorders of ovarian development |
|
47, XXY (Klinefelter Syndrome & variants) |
Disorders in androgen synthesis or action |
Androgen excess |
|
45, X/46, XY (mixed gonadal dysgenesis) |
Other (i.e. hypospadias, congenital HH, cryptorchidism) |
Other (i.e.vaginal or uterine atresia) |
|
46, XX/46, XY (chimerism) |
|
|
Table 3. DSD classification table
The most common DSDs are:
- Klinefelter Syndrome
- Turner Syndrome
- Mixed gonadal dysgenesis
- Congenital adrenal hyperplasia (CAH)
- Androgen insensitivity syndrome
- Cryptorchidism
Klinefelter Syndrome (1 in 500-1000 live births)
Etiology: 47, XXY (classic) vs 46, XY/47, XXY (mosaic)
Symptoms: Tall stature, gynecomastia, small testes, azoospermia, learning difficulties
Diagnosis: High FSH and LH, low AMH and inhibin B, testosterone low in 50-75% of cases
Treatment: Testosterone replacement, spontaneous fertility unlikely
Turner Syndrome (1 in 2500 live births) and mixed gonadal dysgenesis
Etiology: 45 X (classic) vs Mosaic types (or mixed gonadal dysgenesis) like 45, XO/46, XX; 45, XO/46, XY etc.
Symptoms: Lymphedema, webbed neck/low hairline, high-arched palate, shield chest/wide-spaced nipples, short stature, streak ovaries, coarctation of aorta or other CV defects, learning difficulties
Diagnosis: prenatal typically. Based on cardiac anomalies, short stature in childhood, delayed puberty (high FSH & LH) in puberty.
Treatment: Estrogen/progesterone replacement therapy, long-term follow-up (cardiovascular, bone, reproductive health, hearing), spontaneous fertility unlikely, gonadectomy only when Y fragment contains TSPY locus (risk of gonadoblastoma increased)
Congenital Adrenal Hyperplasia (CAH)
Etiology: Group of autosomal recessive disorders, most common type is due to CYP21A2 gene mutations resulting in 21-hydroxylase deficiency. Classic (1:15,000 births; impaired cortisol synthesis). Non-classic (1:100- 1:1,000; cortisol synthesis relatively normal). Less common types include 11-beta hydroxylase deficiency, 17-hydroxylase deficiency, 3-beta- hydroxysteroid dehydrogenase deficiency.
Symptoms: in 21-hydroxylase deficiency, cortisol and aldosterone are not produced therefore patient present with adrenal salt/salt wasting symptoms. Also due to excess ACTH, excessive androgens are produced that can cause virilization of female infants or milder symptoms of hyperandrogenemia (like acne, premature pubarche, irregular periods) in the non-classic form. In 11-beta hydroxylase and 17-hydroxylase deficiency there is HTN due to excess mineralocorticoid and in 3-beta-hds there is adrenal insufficiency and androgen insufficiency, therefore males are under masculinized.
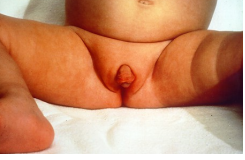
Figure 13 - Female infant with ambiguous genitalia (clitoromegaly, bifid scrotum, rugated folds) due to classic 21-hydroxylase deficiency.
Diagnosis: In 21-hydroxylase deficiency, newborn screen (NBS) detects most cases. If no NBS in place, classic type can present with hyponatremia, hyperkalemia, low aldosterone, high PRA, high 17-hydroxy progesterone, high androstenedione. With the non-classic type there is typically high serum 17-hydroxy progesterone, androstenedione and testosterone.
Management: For classic 21-hydroxylase deficiency: glucocorticoids, mineralocorticoids and also salt for infants. For non-classic 21-hydroxylase deficiency, glucorticoids may be needed but not always (depends on degree of hyperandrogenemia symptoms).
Androgen insensitivity syndrome
Most common cause of male undermasculinization
Etiology: 46, XY/ X-linked recessive/ Androgen Receptor (AR) gene mutation
Symptoms: Wide phenotypic spectrum:
- Complete androgen receptor resistance (CAIS): female phenotype with BL inguinal hernias or with 1o amenorrhea & scanty pubic hair
- Partial androgen receptor resistance (PAIS): ambiguous genitalia with labial masses or male with infertility, decreased pubic hair or beard growth and gynecomastia
Diagnosis: age-appropriate testosterone levels, elevated LH & FSH, normally formed testes, absent/vestigial Mullerian structures, variable presence of Wolffian ducts
Treatment: hormone replacement therapy +/- gonadectomy
Cryptorchidism
Cryptorchidism mean that the testis is not in the scrotum and is not descended by 4 months old. This is the most common congenital abnormality in boys, present in 2-4% of term male infants. Complications include inguinal hernia, testicular torsion or trauma, subfertility, malignant transformation. Management involves orchidopexy (surgically moving the testes into the scrotum) or removal of dysgenic testis. Hormonal therapy (hCG/GnRH) may help testicular descent.

