



Adrenal disorders (cortex and medulla)
The adrenal gland is divided into twodistinct areas, the cortex and the medulla. The cortex is further divided in 3 zones (zona glomerulosa or site of aldosterone synthesis, zona fasciculata or site of cortisol synthesis and zona reticularis or site of androgen synthesis). The medulla serves as the site of catecholamine, metanephrine and normetanephrine synthesis.

Figure 9. Adrenal glandareas and zones with respective hormones synthesized
Cholesterol (low-density lipoprotein or LDL) is the precursor for adrenal steroidogenesis. Adrenal steroidogenesis includes three main pathways, leading to cortisol, aldosterone and androgen synthesis, with androgens eventually being aromatized (converted to estrogens). See figure 10 below, where arrows represent enzymes that convert one substrate to another.
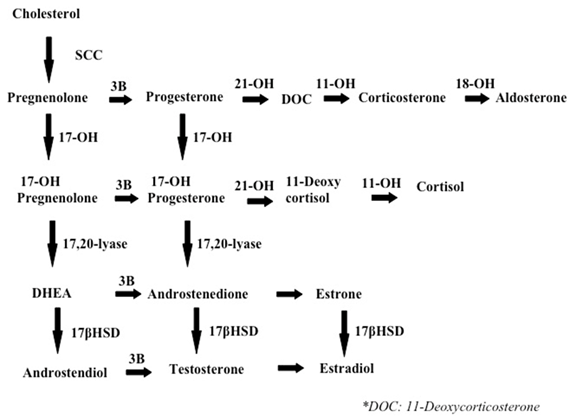
Figure 10: Main pathways of adrenal steriodogenesis
- Cortisol secretion is under the control of the hypothalamic CRH and pituitary ACTH. Cortisol production itself also shuts down the system at the pituitary and hypothalamic levels (negative feedback inhibition). Cortisol actions affect almost every body system (Figure 11 below).
- Aldosterone secretion is under the control of the renin-angiotensin system (not under the control of the brain). Aldosterone regulates water retention, Na and K balance and body's blood pressure.
- Androgens are important for sexual development and reproduction in both males and females. Androgens also serve as precursors of estrogens.
- Catecholamines are neurotransmitters that regulate various central nervous system functions, but are also hormones that regulate cardiovascular and respiratory function, secretions and muscle contraction.
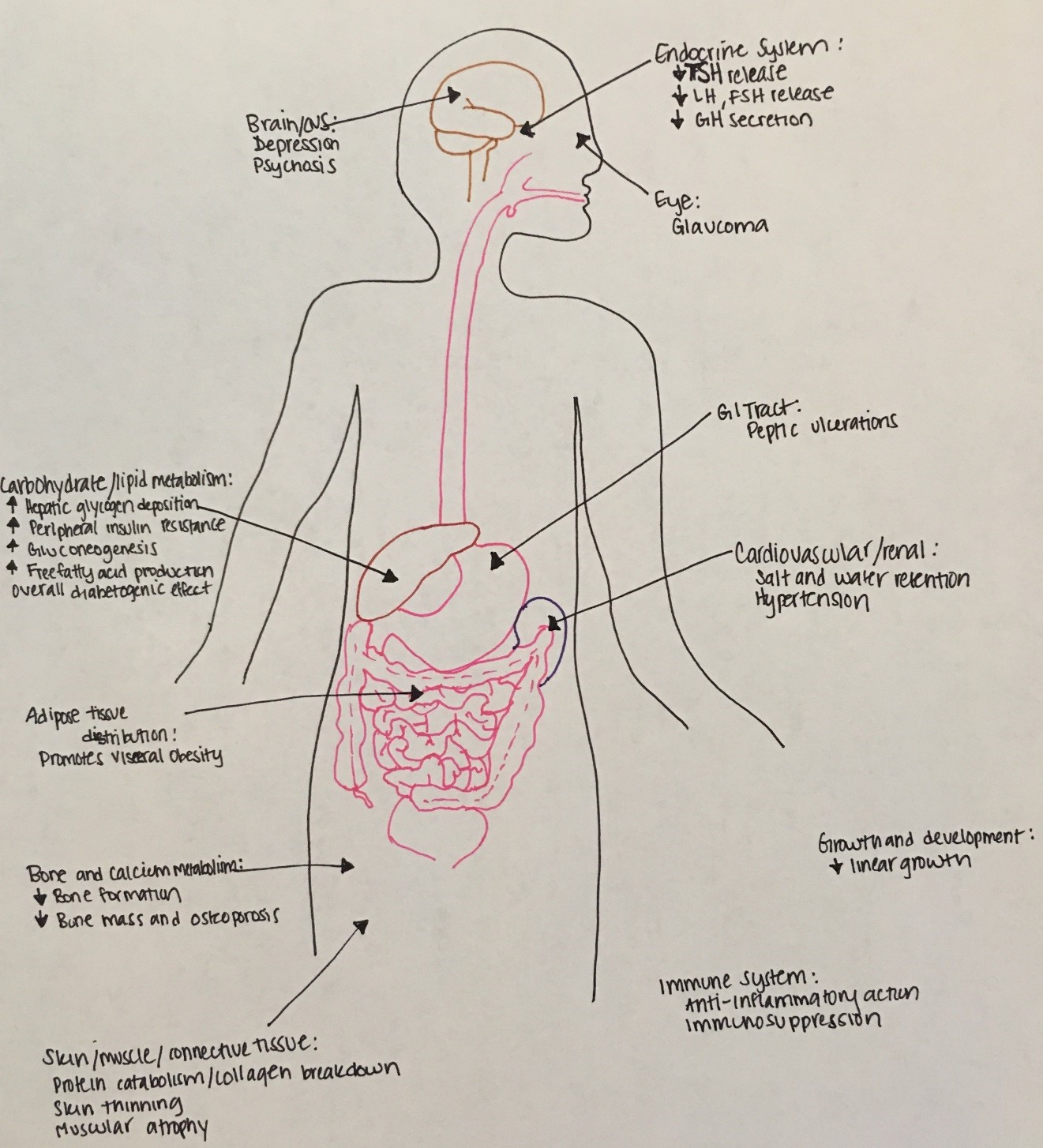
Figure 11: Multiple systemic effects of cortisol
Cortisol excess (or Cushing Syndrome)
Etiology: most commonly exogenous (iatrogenic or due to supraphysiologic exposure to glucocorticoids); rarely endogenous. Endogenous is most commonly due to excess pituitary ACTH secretion (Cushing disease or ACTH producing pituitary adenoma) and rarely due to ectopic ACTH production (e.g. lung or thymic tumor) or excess cortisol secretion by an adrenal tumor.
Symptoms: growth stunting, central obesity, facial plethora, buffalo hump, sleep disorders, hypertension, bruising, tinea corporis, and wide striae
Diagnosis: confirm hypercortisolism with low dose dexamethasone test vs midnight salivary cortisol vs 24-hr urinary free cortisol x3 days, if high cortisol check serum ACTH and refer to endocrinology for more specialized testing and imaging
Treatment: surgical removal of tumor, supplementary radiotherapy for Cushing disease
Adrenal insufficiency
Etiology: most commonly iatrogenic (treatment with suprpaphysiologic doses of glucocorticoids can result to adrenal suppression) vs primary (adrenal gland is malfunctioning) vs central (pituitary or and hypothalamus are malfunctioning). Primary adrenal insufficiency can be autoimmune, due to congenital adrenal hyperplasia, adrenoleukodystrophy, adrenal hemorrhage etc. Central is due to isolated ACTH deficiency or associated with multiple pituitary deficiencies.
Symptoms: hypoglycemia, nausea, emesis, fatigue, anorexia, hypotension, hyponatremia, salt craving and hyperkalemia (if aldosterone deficiency), decreased pubic and axillary hair (if adrenal androgen deficiency), hyperpigmentation (due to high MSH).
Diagnosis: hyponatremia and hyperkalemia, high PRA, low aldosterone, elevated ACTH, failed ACTH stimulation test
Treatment: acute phase (IVF, IV hydrocortisone), chronic phase (glucocorticoids and fludrocortisones)
Congenital adrenal hyperplasia
(discussed in detail under the ambiguous genitalia section)
Pheochromocytoma
Etiology: Catecholamine-secreting tumors, arising from chromaffin cells of the adrenal medulla. Very rare in children and usually inherited (MEN 2b, MEN 2a, NF-1, VHL, familial pheo/para etc).
Symptoms: flushing, sweating, palpitations, hypertension, headaches, emesis, and feeling of impending death
Diagnosis: Plasma or urine fractionated catecholamines/meta- and normetanephrines, CT adrenals and PET-FDG scan
Treatment: Removal of tumor with pre-op preparation to include alpha and beta blockade

